SPRAVATO Nasal spray, solution Ref.[10286] Active ingredients: Esketamine
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Esketamine, the S-enantiomer of racemic ketamine, is a non-selective, non-competitive antagonist of the NmethylD-aspartate (NMDA) receptor, an ionotropic glutamate receptor. The mechanism by which esketamine exerts its antidepressant effect is unknown. The major circulating metabolite of esketamine (noresketamine) demonstrated activity at the same receptor with less affinity.
12.2. Pharmacodynamics
Cardiac Electrophysiology
The effect of SPRAVATO (84 mg nasal spray and 0.8 mg/kg esketamine intravenously infused over 40 minutes) on the QTc interval was evaluated in a randomized, double-blind, placebo-, and positive-controlled (moxifloxacin 400 mg), 4-period, crossover study in 60 healthy subjects. A large increase in heart rate (i.e., >10 bpm) was observed in both intranasal and intravenous esketamine treatment groups. The totality of evidence from the nonclinical and clinical data indicates a lack of clinically relevant QTc prolongation at the therapeutic dose of esketamine.
12.3. Pharmacokinetics
Esketamine exposure increases with dose from 28 mg to 84 mg. The increase in Cmax and AUC values was less than dose-proportional between 28 mg and 56 mg or 84 mg, but it was nearly dose proportional between 56 mg and 84 mg. No accumulation of esketamine in plasma was observed following twice a week administration.
Absorption
The mean absolute bioavailability is approximately 48% following nasal spray administration.
The time to reach maximum esketamine plasma concentration is 20 to 40 minutes after the last nasal spray of a treatment session.
The inter-subject variability of esketamine ranges from 27% to 66% for Cmax and 18% to 45% for AUC∞. The intra-subject variability of esketamine is approximately 15% for Cmax and 10% for AUC∞.
Distribution
The mean steady-state volume of distribution of esketamine administered by the intravenous route is 709 L.
Protein binding of esketamine was approximately 43% to 45%.
The brain-to-plasma ratio of noresketamine is 4- to 6-times lower than that of esketamine.
Elimination
After Cmax was reached following intranasal administration, the decline in plasma esketamine concentrations was biphasic, with rapid decline for the initial 2 to 4 hours and a mean terminal half-life (t1/2) that ranged from 7 to 12 hours. The mean clearance of esketamine is approximately 89 L/hour following intravenous administration. The elimination of the major metabolite, noresketamine, from plasma is slower than esketamine. The decline of noresketamine plasma concentrations is biphasic, with rapid decline for the initial 4 hours and a mean terminal t1/2 of approximately 8 hours.
Metabolism
Esketamine is primarily metabolized to noresketamine metabolite via cytochrome P450 (CYP) enzymes CYP2B6 and CYP3A4 and to a lesser extent CYP2C9 and CYP2C19. Noresketamine is metabolized via CYP-dependent pathways and certain subsequent metabolites undergo glucuronidation.
Excretion
Less than 1% of a dose of nasal esketamine is excreted as unchanged drug in urine. Following intravenous or oral administration, esketamine-derived metabolites were primarily recovered in urine (≥78% of a radiolabeled dose) and to a lesser extent in feces (≤2% of a radiolabeled dose).
Specific Populations
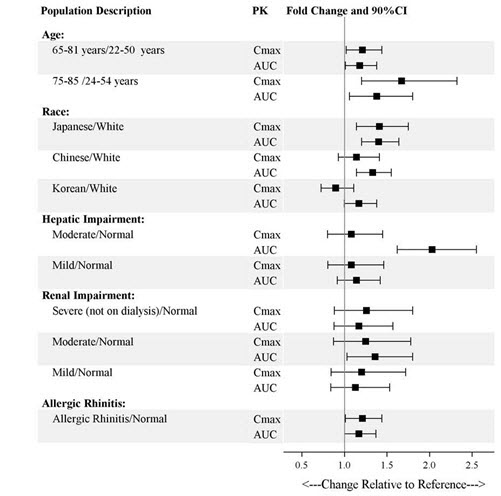
Exposures of esketamine in specific populations are summarized in Figure 1. No significant differences in the pharmacokinetics of SPRAVATO nasal spray were observed for sex and total body weight (>39 to 170 kg) based on population PK analysis. There is no clinical experience with SPRAVATO nasal spray in patients on renal dialysis or with severe (Child-Pugh class C) hepatic impairment.
Figure 1. Effect of Specific Populations on the Pharmacokinetics of Esketamine:
Drug Interaction Studies
The effect of other drugs on the exposures of intranasally administered esketamine are summarized in Figure 2. The effect of SPRAVATO on the exposures of other drugs are summarized in Figure 3. Based on these results, none of the drug-drug interactions are clinically significant.
Figure 2. Effect of Co-administered Drugs on the Pharmacokinetics of Esketamine:

Figure 3. Effect of Esketamine on the Pharmacokinetics of Co-Administered Drugs:

In Vitro Studies
Enzyme Systems: Esketamine has modest induction effects on CYP2B6 and CYP3A4 in human hepatocytes. Esketamine and its major metabolites do not induce CYP1A2. Esketamine and its major circulating metabolites did not show inhibition potential against CYPs and UGTs, except for a weak reversible inhibition of noresketamine on CYP3A4.
Transporter Systems: Esketamine is not a substrate of transporters P-glycoprotein (P-gp; multidrug resistance protein 1), breast cancer resistance protein (BCRP), or organic anion transporter (OATP) 1B1, or OATP1B3. Esketamine and its major circulating metabolites do not inhibit these transporters or multi-drug and toxin extrusion 1 (MATE1) and MATE2-K, or organic cation transporter 2 (OCT2), OAT1, or OAT3.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Once-daily intranasal administration of esketamine at doses equivalent to 4.5, 15, and 45 mg/kg/day (based on a 200-gram rat) did not increase the incidence of tumors in a 2-year rat carcinogenicity study. At the highest dose, the AUC exposure to esketamine was lower than the human exposure (AUC) at the maximum recommended human dose (MRHD) of 84 mg. Once-daily subcutaneous administration of esketamine up to 75 mg/kg/day (reduced to 40 mg/kg/day during week 17) did not increase the incidence of tumors in a 6-month study in transgenic (Tg.rasH2) mice.
Mutagenesis
Esketamine was not mutagenic with or without metabolic activation in the Ames test. Genotoxic effects with esketamine were seen in a screening in vitro micronucleus test in the presence of metabolic activation. However, intravenously-administered esketamine was devoid of genotoxic properties in an in vivo bone marrow micronucleus test in rats and an in vivo Comet assay in rat liver cells.
Impairment of Fertility
Esketamine was administered intranasally to both male and female rats before mating, throughout the mating period, and up to day 7 of gestation at doses equivalent to 4.5, 15, and 45 mg/kg/day (based on a 200-gram rat), which are approximately 0.05, 0.3, and 0.6-times the maximum recommended human dose (MRHD) of 84 mg/day based on mean AUC exposures, respectively. Estrous cycle irregularities were observed at the high dose of 45 mg/kg/day and increased time to mate was observed at doses ≥15 mg/kg/day without an overall effect on mating or fertility indices. The No Observed Adverse Effect Level (NOAEL) for mating and fertility is 45 mg/kg/day which is 0.6 times the esketamine exposures at MRHD of 84 mg/day.
13.2. Animal Toxicology and/or Pharmacology
Neurotoxicity
In a single-dose neuronal toxicity study where esketamine was administered intranasally to adult female rats, there were no findings of neuronal vacuolation in the brain up to an estimated dose equivalent of 45 mg/kg for a 200-gram rat with a safety margin of 1.8 and 4.5 times the clinical exposures for AUC and Cmax, respectively, to the MRHD of 84 mg/day. In a second single-dose neurotoxicity study conducted with intranasally administered esketamine to adult female rats, there were no findings of neuronal necrosis up to a dose equivalent of 270 mg/kg for a 200-gram rat which has a safety margin of 18-fold and 23-fold, respectively, to AUC and Cmax exposures at the MRHD of 84 mg/day. Neuronal vacuolation was not examined in this study.
In a single-dose neuronal toxicity study in adult rats, subcutaneously administered racemic ketamine caused neuronal vacuolation in layer I of the retrosplenial cortex of the brain without neuronal necrosis at a dose of 60 mg/kg. The NOAEL for vacuolation in this study was 15 mg/kg. Estimating 50% of the exposure to be from esketamine, the NOAEL for neuronal vacuolation is 1.6-times and 4.5-times and the NOAEL for neuronal necrosis is 10-times and 16-times exposures, respectively, for AUC and Cmax to the clinical exposure at the MRHD of 84 mg/day. The relevance of these findings to humans is unknown.
14. Clinical Studies
14.1 Treatment-Resistant Depression
Short-Term Study
SPRAVATO was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week), Phase 3 study (Study 1; NCT02418585) in adult patients 18 to <65 years old with treatment-resistant depression (TRD). Patients in Study 1 met DSM-5 criteria for major depressive disorder (MDD) and in the current depressive episode, had not responded adequately to at least two different antidepressants of adequate dose and duration. After discontinuing prior antidepressant treatments, patients in Study 1 were randomized to receive twice weekly doses of intranasal SPRAVATO (flexible dose; 56 mg or 84 mg) or intranasal placebo. All patients also received open-label concomitant treatment with a newly initiated daily oral antidepressant (AD) (duloxetine, escitalopram, sertraline, or extended-release venlafaxine as determined by the investigator based on patient's prior treatment history). SPRAVATO could be titrated up to 84 mg starting with the second dose based on investigator discretion.
The demographic and baseline disease characteristics of patients in Study 1 were similar for the SPRAVATO and placebo nasal spray groups. Patients had a median age of 47 years (range 19 to 64 years) and were 62% female, 93% Caucasian, and 5% Black. The newly initiated oral AD was an SSRI in 32% of patients and an SNRI in 68% of patients.
In Study 1, the primary efficacy measure was change from baseline in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score at the end of the 4-week double-blind induction phase. The MADRS is a ten-item, clinician-rated scale used to assess severity of depressive symptoms. Scores on the MADRS range from 0 to 60, with higher scores indicating more severe depression. SPRAVATO plus a newly initiated oral AD demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus a newly initiated oral AD (see Table 9).
Table 9. Primary Efficacy Results for Change from Baseline in MADRS Total Score at Week 4 in Patients with TRD in Study 1:
| Treatment Group | Number of Patients | Mean Baseline Score (SD) | LS Mean (SE) Change from Baseline to end of Week 4 | LS Mean Difference (95% CI)* |
|---|---|---|---|---|
| SPRAVATO (56 mg or 84 mg) + Oral AD† | 114 | 37.0 (5.7) | -19.8 (1.3) | -4.0 (-7.3; -0.6) |
| Placebo nasal spray + Oral AD | 109 | 37.3 (5.7) | -15.8 (1.3) | - |
SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=confidence interval; AD=antidepressant
* Difference (SPRAVATO + Oral AD minus Placebo nasal spray + Oral AD) in leastsquares mean change from baseline
† SPRAVATO + Oral AD was statistically significantly superior to placebo nasal spray + oral AD
Time Course of Treatment Response:
Figure 4 shows the time course of response for the primary efficacy measure (MADRS) in Study 1. Most of SPRAVATO's treatment difference compared to placebo was observed at 24 hours. Between 24 hours and Day 28, both the SPRAVATO and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 28. At Day 28, 67% of the patients randomized to SPRAVATO were receiving 84 mg twice weekly.
Figure 4. Least Squares Mean Change from Baseline in MADRS Total Score Over Time in Patients with TRD in Study 1* (Full Analysis Set):

* Note: In this flexible-dose study, dosing was individualized based on efficacy and tolerability. Few subjects (<10%) had reduction in SPRAVATO dosage from 84 mg to 56 mg twice weekly.
Treatment-Resistant Depression – Long-term Study
Study 2 (NCT02493868) was a long-term randomized, double-blind, parallel-group, multicenter maintenance-of-effect study in adults 18 to <65 years of age who were known remitters and responders to SPRAVATO. Patients in this study were responders in one of two short-term controlled trials (Study 1 and another 4-week study) or in an open-label direct-enrollment study in which they received flexibly-dosed SPRAVATO (56 mg or 84 mg twice weekly) plus daily oral AD in an initial 4-week phase.
Stable remission was defined as a MADRS total score ≤12 for at least 3 of the last 4 weeks. Stable response was defined as a MADRS total score reduction ≥50% for the last 2 weeks of optimization and not in remission. After at least 16 initial weeks of treatment with SPRAVATO and an oral AD, stable remitters and stable responders were randomized separately to continue intranasal treatment with SPRAVATO or switch to placebo nasal spray, in both cases with continuation of their oral AD. The primary study endpoint was time to relapse in the stable remitter group. Relapse was defined as a MADRS total score ≥22 for 2 consecutive weeks or hospitalization for worsening depression or any other clinically relevant event indicative of relapse.
The demographic and baseline disease characteristics of the two groups were similar. Patients had a median age of 48 years (range 19 to 64 years) and were 66% female, 90% Caucasian, and 4% Black.
Patients in stable remission who continued treatment with SPRAVATO plus oral AD experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on placebo nasal spray plus an oral AD (see Figure 5).
Figure 5. Time to Relapse in Patients with TRD in Stable Remission in Study 2* (Full Analysis Set):

* Note: The estimated hazard ratio (95% CI) of SPRAVATO + Oral AD relative to Placebo nasal spray + Oral AD based on weighted estimates was 0.4 9 (95% CI: 0.29, 0.84). However, the hazard ratio did not appear constant throughout the trial.
Time to relapse was also significantly delayed in the stable responder population. These patients experienced a statistically significantly longer time to relapse of depressive symptoms than patients on placebo nasal spray plus oral AD (see Figure 6).
Figure 6. Time to Relapse in Patients in Stable Response in Patients with TRD in Study 2* (Full Analysis Set):

* Note: The estimated hazard ratio (95% CI) of SPRAVATO + Oral AD relative to Placebo nasal spray + Oral AD based on Cox proportional hazards model was 0.30 (95% CI: 0.16, 0.55).
In Study 2, based on depressive symptomatology, the majority of stable remitters (69%) received every-other-week dosing for the majority of time during the maintenance phase; 23% of stable remitters received weekly dosing. Among stable responders, 34% received every-other-week dosing and 55% received weekly dosing the majority of time during the maintenance phase. Of the patients randomized to SPRAVATO, 39% received the 56 mg dose and 61% received the 84 mg dose.
14.2 Depressive Symptoms in Patients with Major Depressive Disorder with Acute Suicidal Ideation or Behavior
SPRAVATO was evaluated in two identical Phase 3 short-term (4-week) randomized, double-blind, multicenter, placebo-controlled studies, Study 3 (NCT03039192) and Study 4 (NCT03097133), in adults with moderate-to-severe MDD (MADRS total score >28) who had active suicidal ideation and intent. In these studies, patients received treatment with SPRAVATO 84 mg or placebo nasal spray twice-weekly for 4 weeks. After the first dose, a one-time dose reduction to SPRAVATO 56 mg was allowed for patients unable to tolerate the 84 mg dose. All patients received comprehensive standard of care treatment, including an initial inpatient psychiatric hospitalization and a newly initiated or optimized oral antidepressant (AD) (AD monotherapy or AD plus augmentation therapy) as determined by the investigator. After completion of the 4-week treatment period with SPRAVATO/placebo, study follow-up continued through Day 90.
The baseline demographic and disease characteristics of patients in Study 3 and Study 4 were similar between the SPRAVATO plus standard of care or placebo nasal spray plus standard of care treatment groups. The median patient age was 40 years (range 18 to 64 years), 61% were female; 73% Caucasian and 6% Black; and 63% of patients had at least one prior suicide attempt. Prior to entering the study, 92% of the patients were receiving antidepressant therapy. During the study, as part of standard of care treatment, 40% of patients received AD monotherapy, 54% of patients received AD plus augmentation therapy, and 6% received both AD monotherapy/AD plus augmentation therapy.
The primary efficacy measure was the change from baseline in the MADRS total score at 24 hours after first dose (Day 2). In Study 3 and Study 4, SPRAVATO plus standard of care demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus standard of care (see Table 10).
Table 10. Primary Efficacy Results for Change from Baseline in MADRS Total Score at 24 Hours After First Dose (Studies 3 and 4):
| Study No. | Treatment Group* | Number of Patients | Mean Baseline Score (SD) | LS Mean Change from Baseline to 24 hr Post First Dose (SE) | LS Mean Difference (95% CI)† |
|---|---|---|---|---|---|
| Study 3 | SPRAVATO 84 mg + SOC‡ | 111 | 41.3 (5.87) | -15.9 (1.04) | -3.8 (-6.56; -1.09) |
| Placebo nasal spray + SOC | 112 | 41.0 (6.29) | -12.0 (1.02) | - | |
| Study 4 | SPRAVATO 84 mg + SOC‡ | 113 | 39.4 (5.21) | -16.0 (1.02) | -3.9 (-6.60; -1.11) |
| Placebo nasal spray + SOC | 113 | 39.9 (5.76) | -12.2 (1.05) | - |
SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=confidence interval; SOC=standard of care.
* SOC treatment included an initial inpatient psychiatric hospitalization and a newly initiated or optimized oral antidepressant (antidepressant monotherapy or antidepressant monotherapy plus augmentation therapy).
† Difference (SPRAVATO + SOC minus placebo nasal spray + SOC) in least-squares mean change from baseline.
‡ SPRAVATO + SOC were statistically significantly superior to placebo nasal spray + SOC.
The secondary efficacy measure was the change in Clinical Global Impression of Suicidal Severity - Revised (CGI-SS-r) score at 24 hours after first dose (Day 2). The CGI-SS-r is a one-item, clinician-rated assessment used to rate the current severity of a patient's suicidal ideation and behavior. Scores on the CGI-SS-r range from 0 to 6, with higher scores indicating more severe suicidal ideation and behavior. In Study 3 and Study 4, SPRAVATO plus standard of care did not demonstrate superiority compared to placebo nasal spray plus standard of care in improving CGI-SS-r.
Time Course of Treatment Response:
In both Study 3 and Study 4, SPRAVATO's treatment difference compared to placebo was observed starting at 4 hours. Between 4 hours and Day 25, both the SPRAVATO and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 25. Figure 7 depicts time course of the primary efficacy measure of change in MADRS total score from Study 3.
Figure 7. Least Squares Mean Change from Baseline in MADRS Total Score Over Time in Study 3 (Full Analysis Set):

* Note: In Study 3, after the first dose, a one-time dose reduction to SPRAVATO 56 mg was allowed for patients unable to tolerate the 84 mg dose. Approximately 19% of patients had reduction in SPRAVATO dosage from 84 mg to 56 mg twice weekly.
14.3 Special Safety Studies
Effects on Driving
Two studies were conducted to assess the effects of SPRAVATO on driving skills; one study in adult patients with major depressive disorder (Study 5) and one study in healthy subjects (Study 6). On-road driving performance was assessed by the mean standard deviation of the lateral position (SDLP), a measure of driving impairment.
A single-blind, placebo-controlled study in 25 adult patients with major depressive disorder evaluated the effects of a single 84 mg dose of intranasal SPRAVATO on next day driving and the effect of repeated administration of 84 mg of intranasal SPRAVATO on same-day driving performance (Study 5). For the single dose treatment phase, an ethanol-containing beverage was used as a positive control. The SDLP after administration of single 84 mg dose of SPRAVATO nasal spray was similar to placebo 18 hours post-dose. For the multiple dose treatment phase, the SDLP after repeated administration of 84 mg intranasal SPRAVATO was similar to placebo 6 hours post-dose on Day 11, Day 18, and Day 25.
A randomized, double-blind, cross-over, placebo-controlled study in 23 healthy subjects evaluated the effects of a single 84-mg dose of esketamine nasal spray on driving (Study 6). Mirtazapine (30 mg) was used as a positive control. Driving performance was assessed at 8 hours after SPRAVATO or mirtazapine administration. The SDLP 8 hours after SPRAVATO nasal spray administration was similar to placebo. Two subjects discontinued the driving test after receiving SPRAVATO because of a perceived inability to drive after experiencing post-dose adverse reactions; one subject reported pressure behind the eyes and paresthesia of the hands and feet, the other reported headache with light sensitivity and anxiety.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.