MAYZENT Film-coated tablet Ref.[9922] Active ingredients: Siponimod
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Siponimod is an S1P receptor modulator. Siponimod binds with high affinity to S1P receptors 1 and 5. Siponimod blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. The mechanism by which siponimod exerts therapeutic effects in multiple sclerosis is unknown, but may involve reduction of lymphocyte migration into the central nervous system.
12.2. Pharmacodynamics
Immune System
MAYZENT induces a dose-dependent reduction of the peripheral blood lymphocyte count within 6 hours of the first dose, caused by the reversible sequestration of lymphocytes in lymphoid tissues.
With continued daily dosing, the lymphocyte count continues to decrease, reaching a nadir median (90% CI) lymphocyte count of approximately 0.560 (0.271-1.08) cells/nL in a typical CYP2C9*1/*1 or *1/*2, non-Japanese patient, corresponding to 20% to 30% of baseline. Low lymphocyte counts are maintained with chronic daily dosing [see Warnings and Precautions (5.1)].
Lymphocyte counts returned to the normal range in 90% of patients within 10 days of stopping therapy. After stopping MAYZENT treatment, residual lowering effects on peripheral lymphocyte count may persist for up to 3 to 4 weeks after the last dose [see Warnings and Precautions (5.1)].
Heart Rate and Rhythm
MAYZENT causes a transient reduction in heart rate and atrioventricular conduction upon treatment initiation [see Warnings and Precautions (5.3)]. The maximum decline in heart rate is seen in the first 6 hours post dose. Autonomic responses of the heart, including diurnal variation of heart rate and response to exercise, are not affected by siponimod treatment.
A transient, dose-dependent decrease in heart rate was observed during the initial dosing phase of MAYZENT, which plateaued at doses greater than or equal to 5 mg, and bradyarrhythmic events (AV blocks and sinus pauses) were detected at a higher incidence under MAYZENT treatment, compared to placebo.
No second-degree AV blocks of Mobitz type II or higher degree were observed. Most AV blocks and sinus pauses occurred above the recommended dose of 2 mg, with notably higher incidence under non-titrated conditions compared to dose titration conditions [see Dosage and Administration (2.2, 2.3)].
The decrease in heart rate induced by MAYZENT can be reversed by atropine or isoprenaline.
Beta-Blockers
The negative chronotropic effect of coadministration of siponimod and propranolol was evaluated in a dedicated pharmacodynamics (PD)/safety study. The addition of propranolol on top of siponimod at steady-state had less pronounced negative chronotropic effects (less than additive effect) than the addition of siponimod to propranolol at steady state (additive HR effect) [see Drug Interactions (7.3)].
Cardiac Electrophysiology
In a thorough QT study with doses of 2 mg (recommended dose) and 10 mg (five times the recommended dose) siponimod at steady-state, siponimod treatment resulted in a prolongation of QTc, with the maximum mean (upper bound of the two-sided 90% CI) of 7.8 (9.93) ms at 2 mg dose and 7.2 (9.72) ms at 10 mg dose. There was an absence of dose- and exposure-response relationship for QTc effects with the 5-fold dose and exposures achieved by the supratherapeutic dose. No subject had absolute QTcF greater than 480 ms or ΔQTcF greater than 60 ms for siponimod treatment.
Pulmonary Function
Dose-dependent reductions in absolute forced expiratory volume over 1 second were observed in MAYZENT-treated patients and were greater than in patients taking placebo [see Warnings and Precautions (5.4)].
12.3. Pharmacokinetics
Siponimod concentration increases in an apparent dose-proportional manner after multiple once-daily doses of siponimod 0.3 mg to 20 mg. Steady-state plasma concentrations are reached after approximately 6 days of once-daily dosing, and steady-state levels are approximately 2-3-fold greater than the initial dose. An up-titration regimen is used to reach the clinical therapeutic dose of siponimod of 2 mg after 6 days, and 4 additional days of dosing are required to reach the steady-state-plasma concentrations.
Absorption
The time (Tmax) to reach maximum plasma concentrations (Cmax) after oral administration of immediate release oral dosage forms of siponimod was about 4 hours (range, 3-8 hours). Siponimod absorption is extensive (greater than or equal to 70%, based on the amount of radioactivity excreted in urine and the amount of metabolites in feces extrapolated to infinity). The absolute oral bioavailability of siponimod is approximately 84%. After administration of siponimod 2 mg once-daily over 10 days, a mean Cmax of 30.4 ng/mL and mean area under plasma concentration-time curve over dosing interval (AUCtau) of 558 h*ng/mL were observed on Day 10. Steady-state was reached after approximately 6 days of once-daily administration of siponimod.
Food Effect
Food intake resulted in delayed absorption (the median Tmax increased by approximately 2-3 hours). Food intake had no effect on the systemic exposure of siponimod (Cmax and AUC). Therefore, MAYZENT may be taken without regard to meals.
Distribution
Siponimod distributes to body tissues with a moderate mean volume of distribution of 124 L. Siponimod fraction found in plasma is 68% in humans. Animal studies show that siponimod readily crosses the blood-brain-barrier. Protein binding of siponimod is greater than 99.9% in healthy subjects and in hepatic and renal impaired patients.
Elimination
Metabolism
Siponimod is extensively metabolized, mainly via CYP2C9 (79.3%), followed by CYP3A4 (18.5%). The pharmacological activity of the main metabolites M3 and M17 is not expected to contribute to the clinical effect and the safety of siponimod in humans.
Excretion
An apparent systemic clearance (CL/F) of 3.11 L/h was estimated in MS patients. The apparent elimination half-life is approximately 30 hours.
Siponimod is eliminated from the systemic circulation mainly due to metabolism, and subsequent biliary/fecal excretion. Unchanged siponimod was not detected in urine.
Specific Populations
Male and Female Patients
Gender has no influence on siponimod pharmacokinetics (PK).
Racial or Ethnic Groups
The single-dose PK parameters were not different between Japanese and Caucasians healthy subjects, indicating absence of ethnic sensitivity on the PK of siponimod.
Patients with Renal Impairment
No dose adjustments are needed in patients with renal impairment. Mean siponimod half-life and Cmax (total and unbound) were comparable between subjects with severe renal impairment and healthy subjects. Unbound AUCs were only slightly increased (by 33%), compared to healthy subjects, and it is not expected to be clinically significant. The effects of end-stage renal disease or hemodialysis on the PK of siponimod has not been studied. Due to the high plasma protein binding (greater than 99.9%) of siponimod, hemodialysis is not expected to alter the total and unbound siponimod concentration and no dose adjustments are anticipated based on these considerations.
Patients with Hepatic Impairment
No dose adjustments for siponimod are needed in patients with hepatic impairment. The unbound siponimod AUC parameters are 15% and 50% higher in subjects with moderate and severe hepatic impairment, respectively, in comparison with healthy subjects for the 0.25 mg single dose studied. The increased unbound siponimod AUC in subjects with moderate and severe hepatic impairment is not expected to be clinically significant. The mean half-life of siponimod was unchanged in hepatic impairment.
Drug Interaction Studies
Siponimod (and Metabolites M3, M17) as a Causative Agent of Interaction
In vitro investigations indicated that siponimod and its major systemic metabolites M3 and M17 do not show any clinically relevant drug-drug interaction potential at the therapeutic dose of 2 mg once-daily for all investigated CYP enzymes and transporters.
Siponimod as an Object of Interaction
CYP2C9 is polymorphic and the genotype influences the fractional contributions of the two oxidative metabolism pathways to overall elimination. Physiologically based PK modeling indicates a differential CYP2C9 genotype-dependent inhibition and induction of CYP3A4 pathways. With decreased CYP2C9 metabolic activity in the respective genotypes, a larger effect of the CYP3A4 perpetrators on siponimod exposure is anticipated.
Coadministration of Siponimod with CYP2C9 and CYP3A4 Inhibitors
The coadministration of fluconazole (moderate CYP2C9 and CYP3A4 dual inhibitor) 200 mg daily at steady-state and a single dose of siponimod 4 mg in CYP2C9*1/*1 healthy volunteers led to a 2-fold increase in the AUC of siponimod. Mean siponimod terminal half-life was increased by 50%. Fluconazole led to a 2- to 4-fold increase in the AUCtau,ss of siponimod across different CYP2C9 genotypes, according to in silico evaluation [see Drug Interactions (7.5)].
Coadministration of Siponimod with CYP2C9 and CYP3A4 Inducers
The coadministration of siponimod 2 mg daily in the presence of 600 mg daily doses of rifampin (strong CYP3A4 and moderate CYP2C9 dual inducer) decreased siponimod AUCtau,ss and Cmax,ss by 57% and 45%, respectively in CY2C9*1/*1 subjects. Rifampin and efavirenz (moderate CYP3A4 inducer) reduced the AUCtau,ss of siponimod by up to 78% and up to 52%, respectively, across CYP2C9 genotypes, according to in silico evaluation [see Drug Interactions (7.6)].
Oral Contraceptives
The effects of coadministration of siponimod 2 mg and 4 mg (twice the recommended dosage) once daily with a monophasic oral contraceptive (OC) containing 30 mcg ethinyl estradiol and 150 mcg levonorgestrel were assessed in 24 healthy female subjects (18 to 40 years of age; CYP2C9*1/*1 genotype). There were no clinically relevant effects on the PK or PD of the OC. No interaction studies have been performed with OCs containing other progestagens; however, an effect of siponimod on their exposure is not expected.
12.5. Pharmacogenomics
The CYP2C9 genotype has a significant impact on siponimod metabolism. After a single dose of 0.25 mg siponimod, AUCinf and AUClast was approximately 2- and 4-fold higher in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, while there was only a minor increase of Cmax by 21% and 16%, respectively, compared to extensive metabolizers (CYP2C9*1/*1). Mean half-life is prolonged in CYP2C9*2/*3 and CYP2C9*3/*3 carriers (51 hours and 126 hours, respectively).
An apparent systemic clearance (CL/F) of about 3.11 L/h was estimated in CYP2C9 extensive metabolizer (CYP2C9*1/*1 and CYP2C9*1/*2) MS patients after multiple oral administrations of siponimod. Cl/F is 2.5, 1.9, 1.6, and 0.9 L/h in subjects with the CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3, and CYP2C9*3/*3 genotypes respectively. The resultant increase in siponimod AUC was approximatively 25, 61, 91, and 285% higher in CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3, and CYP2C9*3/*3 subjects, respectively, as compared to CYP2C9*1/*1 subjects [see Dosage and Administration (2.1, 2.3) and Contraindications (4)]. As the apparent clearance estimated for CYP2C9*1/*2 subjects is comparable to that of CYP2C9*1/*1 subjects, similar siponimod exposure is expected for both genotypes.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral carcinogenicity studies of siponimod were conducted in mice and rats. In mice administered siponimod (0, 2, 8, or 25 mg/kg/day) for up to 104 weeks, there was an increase in malignant lymphoma in females at all doses and in hemangiosarcoma and combined hemangioma and hemangiosarcoma at all doses in males and females. The lowest dose tested is approximately 5 times the RHD of 2 mg/day, on a body surface area (mg/m 2) basis.
In rats, administration of siponimod (0, 10, 30, or 90 mg/kg/day in males; 0, 3, 10, or 30 mg/kg/day in females) for up to 104 weeks, there was an increase in thyroid follicular cell adenoma and combined thyroid follicular cell adenoma and carcinoma in males at the highest dose tested. These findings are considered secondary to liver enzyme induction in rats and are not considered relevant to humans. Plasma siponimod exposure (AUC) at the highest dose tested is approximately 200 times that in humans at the RHD.
Mutagenesis
Siponimod was negative in a battery of in vitro (Ames, chromosomal aberration in mammalian cells) and in vivo (micronucleus in mouse and rat) assays.
Impairment of Fertility
When siponimod was administered orally (0, 2, 20, or 200 mg/kg) to male rats (mated with untreated females) prior to and throughout the mating period, there was a dose-related increase in precoital interval at all doses. A decrease in implantation sites, an increase in preimplantation loss, and a decrease in the number of viable fetuses were observed at the highest dose tested. The higher no-effect dose for adverse effects on fertility (20 mg/kg) is approximately 100 times the RHD on a mg/m 2 basis.
When siponimod was administered orally (0, 0.1, 0.3, or 1 mg/kg) to female rats (mated with untreated males) prior to and during mating, and continuing to Day 6 of gestation, no effects on fertility were observed up to the highest dose tested (1 mg/kg). Plasma siponimod exposure (AUC) at the highest dose tested is approximately 16 times that in humans at the RHD.
14. Clinical Studies
The efficacy of MAYZENT was demonstrated in Study 1, a randomized, double-blind, parallel-group, placebo-controlled, time-to-event study in patients with secondary progressive multiple sclerosis (SPMS) who had evidence of disability progression in the prior 2 years, no evidence of relapse in 3 months prior to study enrollment, and an Expanded Disability Status Scale (EDSS) score of 3.0-6.5 at study entry (NCT 01665144).
Patients were randomized to receive either once daily MAYZENT 2 mg or placebo, beginning with a dose titration [see Dosage and Administration (2.2)]. Evaluations were performed at screening, every 3 months during the study, and at the time of a suspected relapse. MRI evaluations were performed at screening and every 12 months.
The primary endpoint of the study was the time to 3-month confirmed disability progression (CDP), defined as at least a 1-point increase from baseline in EDSS (0.5-point increase for patients with baseline EDSS of 5.5 or higher) sustained for 3 months. A prespecified hierarchical analysis consisted of the primary endpoint and 2 secondary endpoints, the time to 3-month confirmed worsening of at least 20% from baseline on the timed 25-foot walk test and the change from baseline in T2 lesion volume. Additional endpoints included annualized relapse rate (relapses/year) and MRI measures of inflammatory disease activity.
Study duration was variable for individual patients (median study duration was 21 months, range 1 day-37 months).
Study 1 randomized 1651 patients to either MAYZENT 2 mg (N = 1105) or placebo (N = 546); 82% of MAYZENT-treated patients and 78% of placebo-treated patients completed the study. Median age was 49.0 years, 95% of patients were white, and 60% female. The median disease duration was 16.0 years, and median EDSS score at baseline was 6.0 (56% of patients had ≥6.0 EDSS at baseline); 36% of patients had one or more relapses in the 2 years prior to study entry; 22% of those patients with available imaging had one or more gadolinium-enhancing lesions on their baseline MRI scan; 78% of patients had been previously treated with an MS therapy.
Results are presented in Table 4. MAYZENT was superior to placebo in reducing the risk of confirmed disability progression, based on a time-to-event analysis (hazard ratio 0.79, p<0.0134; see Figure 1). MAYZENT did not significantly delay the time to 20% deterioration in the timed 25-foot walk, compared to placebo. Patients treated with MAYZENT had a 55% relative reduction in annualized relapse rate, compared to patients on placebo (nominal p-value<0.0001). The absolute reduction in the annualized relapse rate was 0.089. Although MAYZENT had a significant effect on disability progression compared to placebo in patients with active SPMS (e.g., SPMS patients with an MS relapse in the 2 years prior to the study), the effect of MAYZENT in patients with non-active SPMS was not statistically significant (see Figure 2).
Table 4. Clinical and MRI Results From Study 1:
| MAYZENT | <>_.PLACEBO | |
|---|---|---|
| Clinical Outcomes | ||
| Proportion of patients with confirmed disability progression1 | 26% | 32% |
| Relative risk reduction | 21% (p = 0.0134)* | |
| Absolute risk Reduction | 6% | |
| Proportion of patients with confirmed worsening in timed 25-foot walk | 40% | 41% |
| p = NS | ||
| Annualized relapse rate2 | 0.071 | 0.160 |
| Relative reduction (%) | 55% (p<0.01)^ | |
| Absolute reduction | 0.089 | |
| p<0.01 ^ | ||
| MRI Endpoints | ||
| Change from baseline in T2 lesion volume (mm³) (95% CI)3 | 184 (54; 314) | 879 (712; 1047) |
| p<0.01^ | ||
All analyses are based on the full analysis set (FAS), which includes all randomized subjects who took at least one dose of study medication.
p-values are two-sided.
1 Defined as an increase of 1.0 point or more from the baseline Expanded Disability Status Scale (EDSS) score for patients with baseline score of 5.5 or less, or 0.5 or more when the baseline score is greater than 5.5. Progression confirmed at 3 months. Cox proportional hazard model.
2 Defined as the average number of confirmed relapses per year (estimated from negative binomial regression model for recurrent events).
3 Adjusted mean averaged over Months 12 and 24.
* Statistically significant.
NS, Not statistically significant.
^ Nominal p value, not corrected for multiple comparisons.
Figure 1. Time to Confirmed Disability Progression Based on EDSS (Study 1):
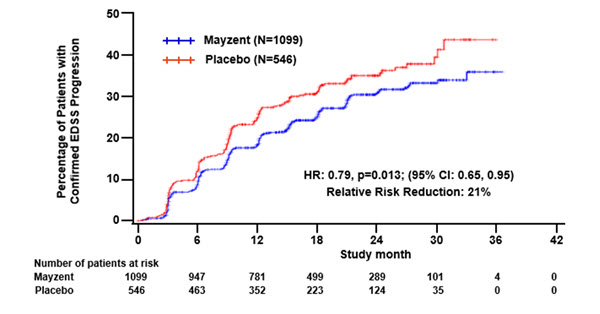
Figure 2. Time to Confirmed Disability Progression Based on EDSS (Study 1), Subgroup Analysis:
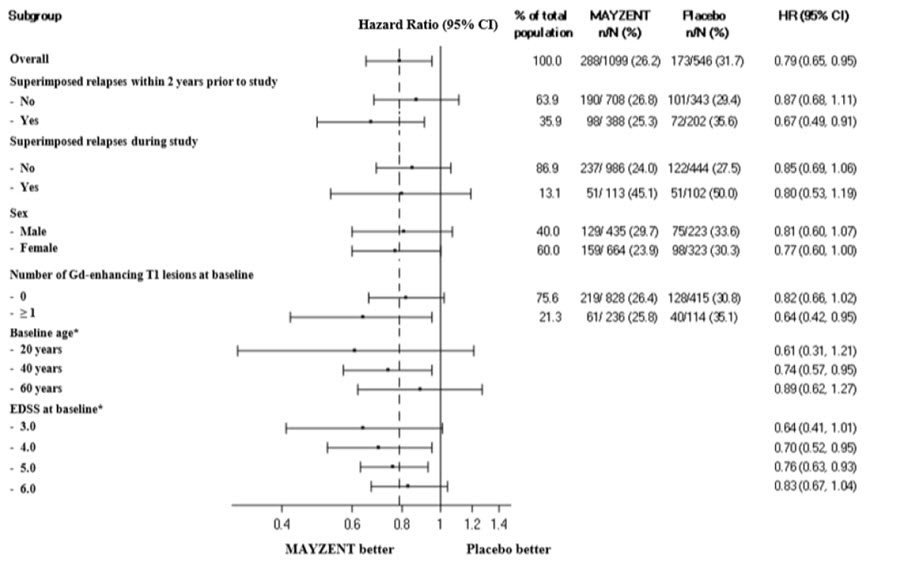
* HR and 95% CI presented are model-based estimates for a range of values of age and EDSS.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.