ABECMA Dispersion for infusion Ref.[49954] Active ingredients: Idecabtagene vicleucel
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents
ATC code: L01XL07
Mechanism of action
Abecma is a chimeric antigen receptor (CAR)-positive T cell therapy targeting B-cell maturation antigen (BCMA), which is expressed on the surface of normal and malignant plasma cells. The CAR construct includes an anti-BCMA scFv-targeting domain for antigen specificity, a transmembrane domain, a CD3-zeta T cell activation domain, and a 4-1BB costimulatory domain. Antigen-specific activation of Abecma results in CAR-positive T cell proliferation, cytokine secretion and subsequent cytolytic killing of BCMA-expressing cells.
Clinical efficacy and safety
KarMMa-3
KarMMa-3 was an open-label, multicentre, randomised, controlled study that evaluated the efficacy and safety of Abecma, compared to standard regimens, in adult patients with relapsed and refractory multiple myeloma who had received two to four prior antimyeloma regimens including an immunomodulatory agent, a proteasome inhibitor, and daratumumab, and were refractory to the most recent prior antimyeloma regimen. A standard regimen was assigned to each patient prior to randomisation, contingent upon the patient's most recent antimyeloma treatment. The standard regimens consisted of daratumumab, pomalidomide, dexamethasone (DPd), daratumumab, bortezomib, dexamethasone (DVd), ixazomib, lenalidomide, dexamethasone (IRd), carfilzomib, dexamethasone (Kd), or elotuzumab, pomalidomide, dexamethasone (EPd). In patients randomised to the Abecma arm, the assigned standard regimen was to be used as bridging therapy, if clinically indicated.
The study included patients who achieved a response (minimal response or better) to at least 1 prior treatment regimen and had ECOG performance status of 0 or 1. The study excluded patients with CNS involvement of myeloma, history of CNS disorders (such as seizures), prior allogeneic SCT or prior treatment with any gene therapy-based therapeutic for cancer, investigational cellular therapy for cancer or BCMA targeted therapy, ongoing treatment with immunosuppresants, serum creatinine clearance <45 mL/min, serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >2.5 times upper limit of normal, and left ventricular ejection fraction (LVEF) <45%. Patients were also excluded if absolute neutrophil count <1000/µL and platelet count <75,000/μL in patients in whom <50% of bone marrow nucleated cells are plasma cells and platelet count <50,000/μL in patients in whom ≥50% of bone marrow nucleated cells are plasma cells.
Patients were randomised 2:1 to receive either Abecma (N=254) or standard regimens (N=132) for relapsed and refractory multiple myeloma. Randomisation was stratified by age, number of prior antimyeloma regimens and high-risk cytogenetics abnormalities. Patients receiving standard regimens were allowed to receive Abecma upon confirmed disease progression.
Patients randomised to Abecma were to receive lymphodepleting chemotherapy consisting of cyclophosphamide (300 mg/m² IV infusion daily for 3 days) and fludarabine (30 mg/m² IV infusion daily for 3 days) starting 5 days prior to the target infusion date of Abecma. Up to 1 cycle of DPd, DVd, IRd, Kd, or EPd anticancer therapy for disease control (bridging therapy) was permitted between apheresis and until 14 days before the start of lymphodepleting chemotherapy.
Of the 254 patients randomised to Abecma, 249 (98%) patients underwent leukapheresis, and 225 (88.6%) patients received Abecma. Of the 225 patients, 192 (85.3%) patients received bridging therapy. Twenty-nine patients did not receive Abecma due to death (n=4), adverse event (n=5), patient withdrawal (n=2), physician decision (n=7), failure to meet lymphodepleting chemotherapy treatment criteria (n=8) or manufacturing failure (n=3).
The allowed dose range was 150 to 540 x 106 CAR-positive T cells. The median actual received dose was 445.3 x 106 CAR-positive T cells (range: 174.9 to 529.0 x 106 CAR-positive T cells). The median time from leukapheresis to product availability was 35 days (range: 24 to 102 days) and the median time from leukapheresis to infusion was 49 days (range: 34 to 117 days).
Of the 132 patients randomised to standard regimens, 126 (95.5%) patients received treatment. Six patients discontinued without receiving treatment due to disease progression (n=1), patient withdrawal (n=3), or physician decision (n=2). Patients receiving standard regimens were allowed to receive Abecma at investigator's request, upon confirmed disease progression by the independent review committee (IRC) based on the International Myeloma Working Group (IMWG) criteria and confirmed eligibility. Of the eligible patients, 69 (54.8%) underwent leukapheresis and 60 (47.6%) received Abecma.
Table 4 summarises the baseline patient and disease characteristics in KarMMa-3 study.
Table 4. Baseline demographic/disease characteristics for patients in KarMMa-3 study:
| Characteristic | Abecma (N=254) | Standard regimens (N=132) |
|---|---|---|
| Age (years) | ||
| Median (min, max) | 63 (30, 81) | 63 (42, 83) |
| ≥65 years, n (%) | 104 (40.9) | 54 (40.9) |
| ≥75 years, n (%) | 12 (4.7) | 9 (6.8) |
| Gender, male, n (%) | 156 (61.4) | 79 (59.8) |
| Race, n (%) | ||
| Asian | 7 (2.8) | 5 (3.8) |
| Black | 18 (7.1) | 18 (13.6) |
| White | 172 (67.7) | 78 (59.1) |
| ECOG performance status, n (%)a | ||
| 0 | 120 (47.2) | 66 (50.0) |
| 1 | 133 (52.4) | 62 (47.0) |
| 2 | 0 | 3 (2.3) |
| 3 | 1 (0.4) | 1 (0.8) |
| Patients with extramedullary plasmacytoma, n (%) | 61 (24.0) | 32 (24.2) |
| Time since initial diagnosis (year) | ||
| n median (min, max) | 251 4.1 (0.6, 21.8) | 131 4.0 (0.7, 17.7) |
| Prior stem cell transplant, n (%) | 214 (84.3) | 114 (86.4) |
| Baseline cytogenetic abnormality, n (%)b | ||
| High riskc | 107 (42.1) | 61 (46.2) |
| Non-high risk | 114 (44.9) | 55 (41.7) |
| Not evaluable/Missing | 33 (13.0) | 16 (12.1) |
| Revised ISS stage at baseline (derived)d, n (%) | ||
| Stage I | 50 (19.7) | 26 (19.7) |
| Stage II | 150 (59.1) | 82 (62.1) |
| Stage III | 31 (12.2) | 14 (10.6) |
| Unknown | 23 (9.1) | 10 (7.6) |
| Distribution of prior anti- myeloma regimens, n (%) | ||
| 2 | 78 (30.7) | 39 (29.5) |
| 3 | 95 (37.4) | 49 (37.1) |
| 4 | 81 (31.9) | 44 (33.3) |
| Refractory status to prior classes of therapy, n (%) | ||
| IMiD | 224 (88.2) | 124 (93.9) |
| Proteasome inhibitor (PI) | 189 (74.4) | 95 (72.0) |
| Anti-CD38 antibodies | 242 (95.3) | 124 (93.9) |
| Triple refractorye, n (%) | 164 (64.6) | 89 (67.4) |
ECOG = Eastern Cooperative Oncology Group; IMiD = immunomodulatory agents; ISS = International Staging System;
max = maximum; min = minimum
a All subjects had ECOG score 0 or 1 at screening, but the ECOG score may be >1 at baseline.
b Baseline cytogenetic abnormality was based on baseline cytogenetics from central laboratory if available. If central laboratory was not available or was unknown, cytogenetics prior to screening was used.
c High-risk defined as deletion in chromosome 17p (del[17p]), translocation involving chromosomes 4 and 14 (t[4;14]) or translocation involving chromosomes 14 and 16 (t[14;16]).
d Revised ISS was derived using baseline ISS stage, cytogenic abnormality and serum lactate dehydrogenase.
e Triple refractory is defined as refractory to an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 antibody.
The primary efficacy endpoint was progression free survival (PFS) according to the IMWG Uniform Response Criteria for Multiple Myeloma as determined by an Independent Review Committee (IRC). Other efficacy measures included overall response rate (ORR), overall survival (OS) and patient-reported outcomes. At a pre-specified interim analysis at 80% information fraction with a median follow up time of 18.6 months, Abecma demonstrated a statistically significant improvement in PFS compared to the standard regimens arm; HR = 0.493 (95% CI: 0.38, 0.65, two-sided p-value <0.0001). The results of the subsequent primary analysis (shown in Table 5 and Figure 1), with a median follow-up time of 30.9 months, were consistent with the interim analysis.
Table 5. Summary of efficacy results from KarMMa-3 (intent-to-treat population):
| Abecma arm (N=254) | Standard regimens arm (N=132) | |
|---|---|---|
| Progression free survival | ||
| Number of events, n (%) | 184 (72.4) | 105 (79.5) |
| Median, months [95% CI]a | 13.8 [11.8, 16.1] | 4.4 [3.4, 5.8] |
| Hazard ratio [95% CI]b | 0.49 [0.38, 0.63] | |
| , Overall response rate | ||
| n (%) | 181 (71.3) | 56 (42.4) |
| 95% CI (%)c | (65.7, 76.8) | (34.0, 50.9) |
| CR or better (sCR+CR) | 111 (43.7) | 7 (5.3) |
| sCR | 103 (40.6) | 6 (4.5) |
| CR | 8 (3.1) | 1 (0.8) |
| VGPR | 45 (17.7) | 15 (11.4) |
| PR | 25 (9.8) | 34 (25.8) |
| DOR if best response is CR | ||
| N | 111 | 7 |
| Median, months [95% CI] | 15.7 [12.1, 22.1] | 24.1 [4.6, NA] |
| DOR if best response is PR | ||
| N | 181 | 56 |
| Median, months [95% CI] | 16.5 [12.0, 19.4] | 9.7 [5.4, 15.5] |
| MRD-negative status by NGS and ≥ CR | ||
| MRD negativity rate, n (%)d | 57 (22.4) | 1 (0.8) |
| 95% CI (%)c | (17.3, 27.6) | (0.0, 2.2) |
CI=confidence interval; CR=complete response; DOR=duration of response; MRD=minimal residual disease; PR=partial response; sCR=stringent complete response; VGPR=very good partial response.
a Kaplan-Meier estimate.
b Based on stratified univariate Cox proportional hazards model.
c Two-sided Wald confidence interval.
d MRD negativity was defined as the proportion of all patients in the ITT population who achieved CR or stringent CR and are MRD negative at any timepoint within 3 months prior to achieving CR or stringent CR until the time of progression or death. Based on a threshold of 10-5 using ClonoSEQ, a next-generation sequencing (NGS) assay.
Figure 1. Kaplan-Meier plot of progression-free survival based on IRC assessment in KarMMa-3 study (intent-to-treat population):

At the time of the final analysis for PFS, 74% of planned OS events were reached. Patients receiving standard regimens were allowed to receive Abecma upon confirmed disease progression; the OS data are therefore confounded by 74 (56.1%) patients from the standard regimen arm who received Abecma as a subsequent therapy. The median OS for Abecma was 41.4 months (95% CI: 30.9, NR) versus standard regimens 37.9 months (95% CI: 23.4, NR); HR = 1.01 (95% CI: 0.73, 1.40). Figure 2 shows the Kaplan-Meier curve for OS in the intent-to-treat population (not corrected for cross-over).
Compared to the standard regimens arm (9/132; 6.8%), a higher proportion of patients experienced death within 6 months after randomisation in the Abecma arm (30/254; 11.8%). Of the 30 patients with an early death event in the Abecma arm, 17 patients never received Abecma treatment, and 13 of these 17 died of disease progression. High-risk factors such as high-risk cytogenetic abnormalities, RISS stage III, presence of extramedullary plasmacytoma or high tumour burden (see section 4.4 on rapidly progressing disease) are associated with higher risk of early death.
Figure 2. Kaplan-Meier plot of overall survival based on IRC assessment in KarMMa-3 study (intent-to-treat population):
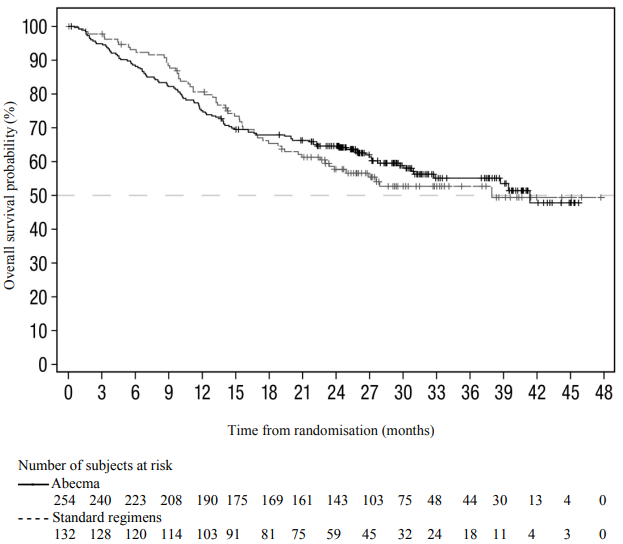
KarMMa
KarMMa was an open-label, single-arm, multicentre study that evaluated the efficacy and safety of Abecma in adult patients with relapsed and refractory multiple myeloma who had received at least 3 prior antimyeloma therapies including an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 antibody and who were refractory to the last treatment regimen. Patients with CNS involvement of myeloma, a history of other BCMA targeting therapies, allogeneic SCT or prior gene therapy based or other genetically modified T cell therapy were excluded. Patients with a history of CNS disorders (such as seizures), inadequate hepatic, renal, bone marrow function, cardiac, pulmonary function or ongoing treatment with immunosuppressants were excluded.
The study consisted of pre-treatment (screening, leukapheresis and bridging therapy [if needed]); treatment (lymphodepleting chemotherapy and Abecma infusion); and posttreatment (ongoing) for a minimum of 24 months following Abecma infusion or until documented disease progression, whichever was longer. The lymphodepleting chemotherapy period was one 3-day cycle of cyclophosphamide (300 mg/m² IV infusion daily for 3 days) and fludarabine (30 mg/m² IV infusion daily for 3 days) starting 5 days prior to the target infusion date of Abecma. Patients were hospitalised for 14 days after infusion of Abecma to monitor and manage potential CRS and neurotoxicity.
Of the 140 patients who were enrolled (i.e. underwent leukapheresis), 128 patients received the Abecma infusion. Out of the 140 patients, only one did not receive the product due to manufacturing failure. Eleven other patients were not treated with Abecma, due to physician decision (n=3), patient withdrawal (n=4), adverse events (n=1), progressive disease (n=1) or death (n=2) prior to receiving Abecma.
Anticancer therapy for disease control (bridging) was permitted between apheresis and lymphodepletion with the last dose being administered at least 14 days prior to initiation of lymphodepleting chemotherapy. Of the 128 patients treated with Abecma, most patients (87.5%) received anticancer therapy for disease control at the discretion of the investigator.
The doses targeted in the clinical study were 150, 300 or 450 x 106 CAR-positive T cells per infusion. The allowed dose range was 150 to 540 x 106 CAR-positive T cells. Table 6 below shows the target dose levels used in the clinical study based on total CAR-positive T cells and the corresponding range of actual dose administered defined as CAR-positive viable T cells.
Table 6. Total CAR-positive T cells dose with the corresponding dose range of CAR-positive viable T cells (x106):
| Target dose based on total CAR-positive T cells, including both viable and non-viable cells (x106) | CAR-positive viable T cells (x106) (min, max) |
|---|---|
| 150 | 133 to 181 |
| 300 | 254 to 299 |
| 450 | 307 to 485 |
Table 7 summarises the baseline patient and disease characteristics for the enrolled and treated population in study.
Table 7. Baseline demographic/disease characteristics for study population:
| Characteristic | Total enrolled (N=140) | Total treated (N=128) |
|---|---|---|
| Age (years) | ||
| Median (min, max) | 60.5 (33, 78) | 60.5 (33, 78) |
| ≥65 years, n (%) | 48 (34.3) | 45 (35.2) |
| ≥75 years, n (%) | 5 (3.6) | 4 (3.1) |
| Gender, male, n (%) | 82 (58.6) | 76 (59.4) |
| Race, n (%) | ||
| Asian | 3 (2.1) | 3 (2.3) |
| Black | 8 (5.7) | 6 (4.7) |
| White | 113 (80.7) | 103 (80.5) |
| ECOG performance status, n (%) | ||
| 0 | 60 (42.9) | 57 (44.5) |
| 1 | 77 (55.0) | 68 (53.1) |
| 2a | 3 (2.1) | 3 (2.3) |
| Patients with extramedullary plasmacytoma, n (%) | 52 (37.1) | 50 (39.1) |
| Time since initial diagnosis (years), median (min, max) | 6 (1.0, 17.9) | 6 (1.0, 17.9) |
| Prior stem cell transplant, n (%) | 131 (93.6) | 120 (93.8) |
| Baseline cytogenetic high riskb,c | 46 (32.9) | 45 (35.2) |
| Revised ISS stage at baseline (derived)d, n (%) | ||
| Stage Ι | 14 (10.0) | 14 (10.9) |
| Stage ΙΙ | 97 (69.3) | 90 (70.3) |
| Stage IIΙ | 26 (18.6) | 21 (16.4) |
| Unknown | 3 (2.1) | 3 (2.3) |
| Number of prior anti-myeloma therapiese, median (min, max) | 6 (3, 17) | 6 (3, 16) |
| Triple refractoryf, n (%) | 117 (83.6) | 108 (84.4) |
| Creatinine clearance (mL/min), n (%) | ||
| <30 | 3 (2.1) | 1 (0.8) |
| 30 to <45 | 9 (6.4) | 8 (6.3) |
| 45 to <60 | 13 (9.3) | 10 (7.8) |
| 60 to <80 | 38 (27.1) | 36 (28.1) |
| ≥80 | 77 (55.0) | 73 (57.0) |
max = maximum; min = minimum
a These patients had ECOG scores of <2 at screening for eligibility but subsequently deteriorated to ECOG scores nt differences in the safety or effectiveness of Abecma were observed between these patients and patientof ≥2 at baseline prior to start of LD chemotherapy.
b Baseline cytogenetic abnormality was based on baseline cytogenetics from central laboratory if available. If central laboratory was not available or was unknown, cytogenetics prior to screening was used.
c High-risk defined as deletion in chromosome 17p (del[17p]), translocation involving chromosomes 4 and 14 (t[4;14]) or translocation involving chromosomes 14 and 16 (t[14;16]).
d Revised ISS was derived using baseline ISS stage, cytogenic abnormality and serum lactate dehydrogenase.
e Induction with or without haematopoietic stem cell transplant and with or without maintenance therapy was considered a single therapy.
f Triple refractory is defined as refractory to an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 antibody.
The median time from leukapheresis to product availability was 32 days (range: 24 to 55 days) and the median time from leukapheresis to infusion was 40 days (range: 33 to 79 days). The median actual dose received across all doses targeted in the clinical study was 315.3 x 106 CAR-positive T cells (range 150.5 to 518.4).
Efficacy was assessed on the basis of overall response rate (ORR), complete response (CR) rate and duration of response (DOR), as determined by an independent review committee. Other efficacy endpoints included minimal residual disease (MRD) using next-generation sequencing (NGS).
Efficacy results across doses targeted in the clinical study (150 to 450 x 106 CAR-positive T cells) are shown in the Table 8. Median follow-up was 19.9 months for all Abecma treated patients.
Table 8. Summary of efficacy based on the KarMMa study:
| Enrolleda (N=140) | Treated population Target dose of Abecma (CAR-positive T cells) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 150 x 106b (N=4) | 300 x 106 (N=70) | 450 x 106 (N=54) | Total 150 to 450 x 106 (N=128) | |||||||||
| Overall response rate (sCR+CR+VGPR+PR), n (%) | 94 (67.1) | 2 (50.0) | 48 (68.6) | 44 (81.5) | 94 (73.4) | |||||||
| 95% CIc | 59.4, 74.9 | 6.8, 93.2 | 56.4, 79.1 | 68.6, 90.7 | 65.8, 81.1 | |||||||
| CR or better, n (%) | 42 (30.0) | 1 (25.0) | 20 (28.6) | 21 (38.9) | 42 (32.8) | |||||||
| 95% CIc | 22.4, 37.6 | 0.6, 80.6 | 18.4, 40.6 | 25.9, 53.1 | 24.7, 40.9 | |||||||
| VGPR or better, n (%) | 68 (48.6) | 2 (50.0) | 31 (44.3) | 35 (64.8) | 68 (53.1) | |||||||
| 95% CIc | 40.3, 56.9 | 6.8, 93.2 | 32.4, 56.7 | 50.6, 77.3 | 44.5, 61.8 | |||||||
| MRD-negative statusd and ≥ CR | ||||||||||||
| Based on treated patients | – | 4 | 70 | 54 | 128 | |||||||
| n (%) | – | 1 (25.0) | 17 (24.3) | 14 (25.9) | 32 (25.0) | |||||||
| 95% CI | – | 0.6, 80.6 | 14.8, 36.0 | 15.0, 39.7 | 17.8, 33.4 | |||||||
| Time to response, n | 94 | 2 | 48 | 44 | 94 | |||||||
| Median (months) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | |||||||
| Min, max | 0.5, 8.8 | 1.0, 1.0 | 0.5, 8.8 | 0.9, 2.0 | 0.5, 8.8 | Duration of response (PR or better)e, n | 94 | 2 | 48 | 44 | 94 | |
| Median (months) | 10.6 | 15.8 | 8.5 | 11.3 | 10.6 | |||||||
| 95% CI | 8.0, 11.4 | 2.8, 28.8 | 5.4, 11.0 | 10.3, 17.0 | 8.0, 11.4 | |||||||
NE = not estimable; PR = partial response; sCR = stringent complete response; VGPR = very good partial response.
a All patients who underwent leukapheresis.
b The 150 x 106 CAR-positive T cell dose is not part of the approved dose range.
c For "Total (Treated population" and "Enrolled population"): Wald CI; for individual target dose levels: Clopper-Pearson exact CI.
d Based on a threshold of 10-5 using a next-generation sequencing assay. 95% CI for percentage of MRD negativity use Clopper-Pearson exact CI for individual target dose levels as well as for Treated population.
e Median and 95% CI are based on the Kaplan-Meier approach.
Note: The target dose is 450 x 106 CAR-positive T cells within a range of 150 to 540 × 106 CAR-positive T cells. The 150 x 106 CAR-positive T cell dose is not part of the approved dose range.
The Kaplan-Meier curve of duration of response by best overall response is shown in Figure 3.
Figure 3. Kaplan-Meier curve of duration of response based on independent response committee review according to IMWG criteria – by best overall response (Abecma-treated population):
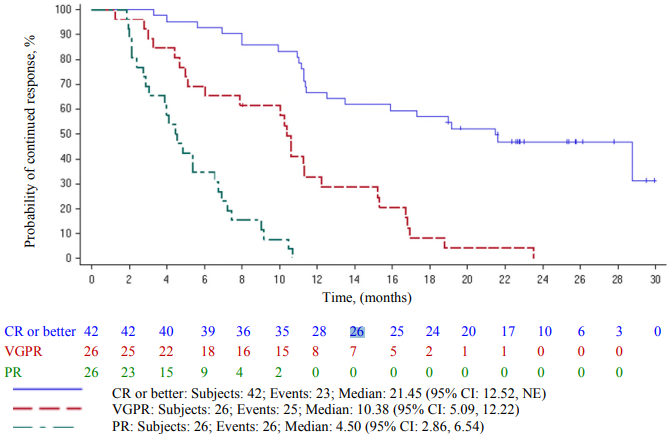
CI = confidence interval; IMWG = International Myeloma Working Group; NE = not estimable. Two patients with 150 x 106 CAR-positive T cell dose, which is not part of the approved dose range, are included in Figure 3.
Special populations
Elderly
In the clinical trial of Abecma, 48 (34.3%) patients in the KarMMa study were 65 years of age or older and 5 (3.6%) were 75 years of age or older (see Table 5). No clinically important differences in the safety or effectiveness of Abecma were observed between these patients and patients younger than 65 years of age.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Abecma in all subsets of the paediatric population in the treatment of mature B-cell neoplasms (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called 'conditional approval scheme'. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Following Abecma infusion, the CAR-positive T cells proliferate and undergo rapid multi-log expansion followed by a bi-exponential decline. The median time of maximal expansion in peripheral blood (Tmax) occurred 11 days after infusion.
Abecma can persist in peripheral blood for up to 1 year post-infusion.
Abecma transgene levels were positively associated with objective tumour response (partial response or better). In patients who received Abecma in the KarMMa-3 study, the median Cmax levels in responders (N=180) were approximately 5.4-fold higher compared to the corresponding levels in non-responders (N=40). Median AUC0-28days in responders (N=180) was approximately 5.5-fold higher than non-responders (N=38). In patients who received Abecma in the KarMMa study, the median Cmax levels in responders (N=93) were approximately 4.5-fold higher compared to the corresponding levels in non-responders (N=34). Median AUC0-28days in responding patients (N=93) was approximately 5.5-fold higher than non-responders (N=32).
Special populations
Renal and hepatic impairment
Hepatic and renal impairment studies of Abecma were not conducted.
Effects of age, weight, gender or race
Age (range: 30 to 81 years) had no impact on Abecma expansion parameters. The pharmacokinetics of Abecma in patients less than 18 years of age have not been evaluated.
Patients with lower body weight had higher cellular expansion. Due to high variability in pharmacokinetic cellular expansion, the overall effect of weight on the expanison parameters of Abecma is considered not to be clinically relevant.
Gender had no impact on Abecma expansion parameters.
Race and ethnicity had no significant impact on Abecma expansion parameters.
5.3. Preclinical safety data
Abecma comprises engineered human T cells, therefore there are no representative in vitro assays, ex vivo models, or in vivo models that can accurately address the toxicological characteristics of the human product. Hence, traditional toxicology studies used for drug development were not performed. Genotoxicity assays and carcinogenicity studies were not conducted.
In vitro expansion studies from healthy donors and patients showed no evidence for transformation and/or immortalisation and no preferential integration near genes of concern in Abecma T cells.
Given the nature of the product, non-clinical studies on fertility, reproduction and development were not conducted.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.