ADCETRIS Powder for concentrate for solution Ref.[6593] Active ingredients: Brentuximab vedotin
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Takeda Pharma A/S, Delta Park 45, 2665 Vallensbaek Strand, Denmark, medinfoEMEA@takeda.com
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies and antibody drug conjugates
ATC code: L01FX05
Mechanism of action
Brentuximab vedotin is an ADC that delivers an antineoplastic agent that results in apoptotic cell death selectively in CD30-expressing tumour cells. Nonclinical data suggest that the biological activity of brentuximab vedotin results from a multi-step process. Binding of the ADC to CD30 on the cell surface initiates internalisation of the ADC-CD30 complex, which then traffics to the lysosomal compartment. Within the cell, a single defined active species, MMAE, is released via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within the cell, induces cell cycle arrest and results in apoptotic death of the CD30-expressing tumour cell.
Classical HL, sALCL and subtypes of CTCL (including MF and pcALCL) express CD30 as an antigen on the surface of their malignant cells. This expression is independent of disease stage, line of therapy or transplant status. These features make CD30 a target for therapeutic intervention. Because of the CD30-targeted mechanism of action brentuximab vedotin is able to overcome chemo-resistance as CD30 is consistently expressed in patients who are refractory to multi-agent chemotherapy, irrespective of prior transplant status. The CD30-targeted mechanism of action of brentuximab vedotin, the consistent expression of CD30 throughout the classical HL, sALCL and CD30+ CTCL disease and therapeutic spectrums and clinical evidence in CD30-positive malignancies following multiple lines of treatment provide a biologic rationale for its use in patients with relapsed and refractory classical HL, sALCL with or without prior ASCT and CD30+ CTCL after at least 1 prior systemic therapy.
Contributions to the mechanism of action by other antibody associated functions have not been excluded.
Pharmacodynamic effects
Cardiac electrophysiology
Forty-six (46) patients with CD30-expressing haematologic malignancies were evaluable of the 52 patients who received 1.8 mg/kg of brentuximab vedotin every 3 weeks as part of a phase 1, single-arm, open-label, multicenter cardiac safety study. The primary objective was to evaluate the effect of brentuximab vedotin on cardiac ventricular re-polarization and the predefined primary analysis was the change in QTc from baseline to multiple time points in Cycle 1.
The upper 90% confidence interval (CI) around the mean effect on QTc was <10 msec at each of the Cycle 1 and Cycle 3 post-baseline time points. These data indicate the absence of clinically relevant QT prolongation due to brentuximab vedotin administered at a dose of 1.8 mg/kg every 3 weeks in patients with CD30-expressing malignancies.
Clinical efficacy and safety
Hodgkin lymphoma
Study C25003
The efficacy and safety of ADCETRIS were evaluated in a randomised, open-label, 2-arm, multicenter trial in 1334 patients with previously untreated advanced HL in combination with chemotherapy (doxorubicin [A], vinblastine [V] and dacarbazine [D] [AVD]). Patients with nodular lymphocyte predominant HL (NLPHL) were excluded from the study. All patients had a histologically confirmed CD30-expressing disease. Sixty-two percent of patients had extranodal site involvement. Of the 1334 patients, 664 patients were randomised to the ADCETRIS + AVD arm and 670 patients were randomised to the ABVD (doxorubicin [A], bleomycin [B], vinblastine [V] and dacarbazine [D]) arm and stratified by number of International Prognostic Factor Project (IPFP) risk factors and region. Patients were treated on days 1 and 15 of each 28-day cycle with 1.2 mg/kg of ADCETRIS administered as an intravenous infusion over 30 minutes + doxorubicin 25 mg/m², vinblastine 6 mg/m², and dacarbazine 375 mg/m². The median number of cycles received was 6 19 (range, 1 to 6 cycles). Table 6 provides a summary of the baseline patient and disease characteristics. There were no relevant differences in the patient and disease characteristics between the two arms.
Table 6. Summary of baseline patient and disease characteristics in the phase 3 previously untreated HL study:
| Patient Characteristics | ADCETRIS + AVD n=664 | ABVD n=670 |
| Median age (range) | 35 years (18-82) | 37 years (18-83) |
| Patients ≥65 years old n (%) | 60 (9) | 62 (9) |
| Gender, n (%) | 378M (57%) 286F (43%) | 398M (59%) 272F (41%) |
| ECOG status, n (%) | ||
| 0 | 376 (57) | 378 (57) |
| 1 | 260 (39) | 263 (39) |
| 2 | 28 (4) | 27 (4) |
| Missing | 0 | 2 |
| Disease Characteristics | ||
| Median time from HL diagnosis to first dose (range) | 0.92 mo (0.1-21.4) | 0.89 mo (0.0-81.4) |
| Disease stagea at initial diagnosis of HL, n (%) | ||
| III | 237 (36) | 246 (37) |
| IV | 425 (64) | 421 (63) |
| Not applicable | 1 (<1) | 1 (<1) |
| Missing | 0 | 2 (<1) |
| Extranodal involvement at time of diagnosis, n (%) | 411 (62) | 416 (62) |
| IPFPb risk factors, n (%) | ||
| 0-1 | 141 (21) | 141 (21) |
| 2-3 | 354 (53) | 351 (52) |
| 4-7 | 169 (25) | 178 (27) |
| Bone marrow involvement at time of diagnosis or study entry, n (%) | 147 (22) | 151 (23) |
| B symptomsa n (%) | 400 (60) | 381 (57) |
a Per Ann Arbor Staging.
b IPFP = International Prognostic Factor Project.
The primary endpoint in Study C25003 was modified PFS (mPFS) per independent review facility (IRF), defined as time from randomisation to disease progression, death, or evidence of non-complete response (non-CR) after completion of first-line therapy per IRF followed by subsequent anticancer therapy. Timing of the modified event was the date of the first PET scan post completion of first-line therapy demonstrating the absence of complete response (CR), defined as Deauville score of ≥3. The median modified PFS by IRF assessment was not reached in either treatment arm. The results in the intent-to-treat (ITT) population showed a statistically significant improvement in modified PFS for ADCETRIS+ AVD, with a stratified hazard ratio of 0.770 (95% CI, 0.603; 0.983, p=0.035), indicating a 23% reduction in the risk of modified PFS events for ADCETRIS+ AVD versus ABVD.
Table 7 provides the efficacy results for modified PFS and overall survival (OS) in the ITT population.
Table 7. Efficacy results for previously untreated HL patients treated with 1.2 mg/kg of ADCETRIS + AVD on days 1 and 15 of a 28-day cycle (ITT):
| Intent to Treat (ITT) Population | |||
| ADCETRIS + AVD n=664 | ABVD n=670 | Stratified Hazard Ratio and p-value | |
| Number of events (%) | 117 (18) | 146 (22) | 0.77 (95% CI [0.60, 0.98]) p-value = 0.035 |
| Estimated mPFSa per IRF at 2 Year (%) | 82,1 (95% CI [78.8, 85.0]) | 77.2 (95% CI [73.7, 80.4]) | |
| Overall Survivalb Number of deaths (%) | 28 (4) | 39 (6) | 0,73 (95% CI [0.45, 1.18]) p-value = 0.199 |
Figure 1. Modified progression-free survival per IRF in the ITT population (ADCETRIS + AVD vs. ABVD):

Other secondary efficacy endpoints including CR rate and ORR at the end of randomisation regimen, CR rate at the end of first-line therapy, and the rate of PET negativity at the end of Cycle 2, duration of response (DOR), duration of complete remission (DOCR), disease-free survival (DFS) and event-free survival (EFS) all trended in favour of ADCETRIS + AVD in the ITT population.
Pre‑specified subgroup analyses of modified PFS per IRF showed no clinically meaningful difference between the two treatment arms in the elderly population (patients ≥60 years of age [n=186] [HR=1.00, 95% CI (0.58, 1.72)] and ≥65 years of age [n=122] [HR=1.01, 95% CI (0.53, 1.94)]) and patients without extranodal sites (n=445) (HR=1.04, 95% CI [0.67, 1.62]).
As of a 01 June 2021 cut-off date, approximately 5 years after enrollment of the last patient, the results in the ITT population showed a statistically significant improvement in OS in the ADCETRIS + AVD arm compared with patients treated with ABVD [HR=0.59, 95% CI (0.396, 0.879)], see Figure 2.
In the Stage III population, OS results indicated a 14% reduction in the risk of death in the A+AVD arm compared with patients in the ABVD arm (HR=0.86, 95% CI [0.452, 1.648]); in the Stage IV population there was a 52% reduction in the risk of death (HR=0.48, 95% CI [0.286, 0.799]). A subgroup analysis of OS showed no clinically meaningful difference between the two treatment arms for patients without extranodal sites (n=445) (HR=1.18, 95% CI [0.641, 2.187]).
Median OS was not reached for either A+AVD or ABVD patients [95%◦CI (NE, NE)].
Figure 2. Overall survival (ADCETRIS + AVD vs. ABVD) (ITT, 6 years median follow up):

A descriptive analysis of OS was performed using data with median follow-up of over 7 years for OS. In the ITT population, a lower proportion of patients randomized to A + AVD had died (44 deaths, 7%) compared with patients randomized to ABVD (67 deaths, 10%; HR=0.61, 95% CI [0.414, 0.892]). Similar proportions of Stage III patients randomized to A+AVD (20 deaths, 8%) and ABVD (20 deaths, 8%) had died (HR=1.00, 95% CI [0.540, 1.866]). A lower proportion of Stage IV patients randomized to A + AVD (24 deaths, 6%) had died compared with patients randomized to ABVD (46 deaths, 11%; HR=0.48, 95% CI [0.291, 0.784]).
In the ITT population, 33% fewer patients treated with ADCETRIS + AVD in the ITT population received subsequent salvage chemotherapy (n=66) and high-dose chemotherapy and transplant (n=36) compared with those treated with ABVD (n=99 and n=54, respectively). In the Stage IV population, 35% fewer patients treated with ADCETRIS + AVD received subsequent salvage chemotherapy (n=45) compared with those treated with ABVD (n=69) and 22% fewer patients treated with ADCETRIS + AVD received high-dose chemotherapy and transplant (n=29) compared with those treated with ABVD (n=37).
Study SGN35-005
The efficacy and safety of ADCETRIS were evaluated in a randomised, double-blind, placebo-controlled, 2-arm multicenter trial in 329 patients with HL at risk of relapse or progression following ASCT. Patients with known cerebral/meningeal disease, including history of PML were excluded from the study. See Table 8 for patient characteristics. Of the 329 patients, 165 patients were randomised to the treatment arm and 164 patients were randomised to the placebo arm. In the study, patients were to receive their first dose after recovery from ASCT (between days 30-45 following ASCT). Patients were treated with 1.8 mg/kg of ADCETRIS or matching placebo intravenously over 30 minutes every 3 weeks for up to 16 cycles.
Eligible patients were required to have at least one of the following risk factors:
- HL that was refractory to frontline treatment
- Relapsed or progressive HL that occurred <12 months from the end of frontline treatment
- Extranodal involvement at time of pre-ASCT relapse, including extranodal extension of nodal masses into adjacent vital organs.
Table 8. Summary of baseline patient and disease characteristics in the phase 3 HL post-ASCT Study:
| Patient characteristics | ADCETRIS n=165 | Placebo n=164 |
| Median age, years (range) | 33 years (18-71) | 32 years (18-76) |
| Gender | 76M (46%) / 89F (54%) | 97M (59%) / 67F (41%) |
| ECOG status | ||
| 0 | 87 (53%) | 97 (59%) |
| 1 | 77 (47%) | 67 (41%) |
| 2 | 1 (1%) | 0 |
| Disease characteristics | ||
| Median number of prior chemotherapy regimens (range) | 2 (2-8) | 2 (2-7) |
| Median time from HL diagnosis to first dose (range) | 18,7 mo (6.1-204.0) | 18.8 mo (7.4-180.8) |
| Disease stage at initial diagnosis of HL | ||
| Stage I | 1 (1%) | 5 (3%) |
| Stage II | 73 (44%) | 61 (37%) |
| Stage III | 48 (29%) | 45 (27%) |
| Stage IV | 43 (26%) | 51 (31%) |
| Unknown | 0 | 2 (1%) |
| PET scan Status prior to ASCT | ||
| FDG-AVID | 64 (39%) | 51 (31%) |
| FDG-NEGATIVE | 56 (34%) | 57 (35%) |
| NOT DONE | 45 (27%) | 56 (34%) |
| Extranodal involvement at time of pre-ASCT relapse | 54 (33%) | 53 (32%) |
| B symptomsa | 47 (28%) | 40 (24%) |
| Best response to salvage therapy pre-ASCTb | ||
| Complete Response | 61 (37%) | 62 (38%) |
| Partial Response | 57 (35%) | 56 (34%) |
| Stable Disease | 47 (28%) | 46 (28%) |
| HL Status after the end of frontline standard chemotherapyb | ||
| Refractory | 99 (60%) | 97 (59%) |
| Relapse occurred <12 months | 53 (32%) | 54 (33%) |
| Relapse occurred ≥12 months | 13 (8%) | 13 (8%) |
a For refractory disease, or upon progression or relapse after frontline therapy.
b Stratification factors at randomisation.
Efficacy results as of the primary analysis of the primary endpoint are shown in Table 9. The primary endpoint of PFS per IRF was met and showed a difference in median PFS of 18.8 months in favour of the treatment arm.
Table 9. Efficacy results in HL patients at increased risk of relapse or progression following ASCT treated with 1.8 mg/kg of ADCETRIS every 3 weeks (ITT, primary analysis):
| ADCETRIS n=165 | Placebo n=164 | Stratified Hazard Ratio | |
|---|---|---|---|
| Progression Free Survivala | Median per IRF | ||
| 42.9 months (95% CI [30.4, 42.9]) | 24.1 months (95% CI [11.5, -]) | 0.57 (95% CI [0.40, 0.81]) Stratified log-rank test p=0.001 | |
| Median per Investigator | |||
| Not reached (95% CI [26.4, -]) | 15.8 months (95% CI [8.5, -]) | 0.5 (95% CI [0.36, 0,70])b | |
| Overall Survival | Number of deaths (%) | ||
| 28 (17) | 25 (15) | 1.15 (95% CI [0.67, 1.97] | |
a At the time of the primary analysis, the median follow-up time for both arms was 30 months (range, 0 to 50).
b Stratified log-rank test was not performed for PFS per Investigator.
Pre-specified subgroup analyses of PFS per IRF were performed by patients' best response to pre-ASCT salvage therapy, HL status after frontline therapy, age, gender, baseline weight, baseline ECOG performance status, number of treatments pre-ASCT, geographic region, pre-ASCT PET status, B symptom status after failure of frontline therapy, and pre-ASCT extranodal disease status. The analyses showed a consistent trend towards benefit for patients who received ADCETRIS compared with patients who received placebo with the exception of patients ≥65 years of age (n=8).
No differences were observed in quality of life between the treatment and placebo arms. Medical resource utilization (MRU) analysis showed that hospitalizations and outpatient visits, as well as working days/other activities missed by patients and caregivers were lower with ADCETRIS compared with placebo in patients with HL at increased risk of relapse.
An updated analysis conducted after 3 years of follow-up showed a sustained PFS improvement per IRF (HR=0.58 [95% CI (0.41, 0.81)]).
As of study closure, approximately 10 years after enrollment of the first patient, PFS per investigator continued to show a benefit (HR = 0.51 [95% CI (0.37, 0.71)]). Overall survival results were consistent with those reported at the time of primary analysis (HR=1.11 [95% CI (0.72, 1.70)]). Figure 3 shows PFS per investigator in the ITT population as of study closure.
Figure 3. Kaplan-Meier Plot of PFS per investigator (ITT, study closure):

Post-hoc Risk Factor Analyses
Post-hoc analyses were performed for the primary analysis of the primary endpoint to evaluate the impact of increased risk (number of risk factors) on clinical benefit (Table 10). Representative risk factors for these analyses were:
- HL that occurred <12 months or HL that was refractory to frontline therapy
- Best response of PR or SD to most recent salvage therapy as determined by CT and/or PET scanning
- Extranodal disease at pre-ASCT relapse
- B symptoms at pre-ASCT relapse
- Two or more prior salvage therapies.
The results of these post-hoc analyses suggest increased clinical benefit for patients with two or more risk factors but no difference based on any of the individual risk factors. No benefit in terms of PFS or OS has been observed in patients with one risk factor for relapse or progression.
Table 10. Summary of PFS per IRF and OS by number of risk factors in the phase 3 HL post-ASCT Study (primary analysis):
| Progression Free Survival per IRF | ||||||
| Number of Risk Factors = 1 | Number of Risk Factors ≥ 2 | Number of Risk Factors ≥ 3 | ||||
| ADCETRIS n=21 | Placebo n=28 | ADCETRIS n=144 | Placebo n=136 | ADCETRIS n=82 | Placebo n=84 | |
| Number of patients with disease progression or deatha (%) | 9 (43) | 7 (25) | 51 (35) | 68 (50) | 32 (39) | 49 (58) |
| Stratified Hazard Ratio | 1,65 (95% CI [0,60, 4,55])b | 0.49 (95% CI [0.34, 0.71]) | 0.43 (95% CI [0.27, 0.68]) | |||
| Overall Survival | ||||||
| Number of Risk Factors = 1 | Number of Risk Factors ≥ 2 | Number of Risk Factors ≥ 3 | ||||
| ADCETRIS n=21 | Placebo n=28 | ADCETRIS n=144 | Placebo n=136 | ADCETRIS n=82 | Placebo n=84 | |
| Number of deathsc (%) | 5 (24) | 1 (4) | 23 (16) | 24 (18) | 15 (18) | 16 (19) |
| Stratified Hazard Ratio | 7,94 (95% CI [0,93, 68,06])b | 0.94 (95% CI [0.53, 1.67]) | 0.92 (95% CI [0.45, 1.88]) | |||
a Death without either prior progression or more than one missed assessment visit.
b Indicates results from non-stratified analysis.
c Events are death due to any cause.
At the time of the updated analysis (3 years of follow-up) for patients with 2 or more risk factors, the hazard ratio for PFS per IRF was 0.49 (95% CI [0.34, 0.71]) and the hazard ratio for PFS per investigator was 0.41 (95% CI [0.29, 0.58]) (see Figures 4 and 5).
Figure 4. Kaplan-Meier Plot of PFS per IRF in Patients with ≥2 Risk Factors (3-year follow-up):

Figure 5. Kaplan-Meier Plot of PFS per Investigator in Patients with ≥2 Risk Factors (3-year follow-up):

As of study closure, approximately 10 years after enrollment of the first patient, the hazard ratio for PFS per investigator for patients with 2 or more risk factors was 0.41 (95% CI [0.29, 0.58]). The hazard ratio for PFS per investigator for patients with 3 or more risk factors was 0.38 (95% CI [0.25, 0.59]). Overall survival results remained consistent with those observed as of the primary analysis.
Study SG035-0003
The efficacy and safety of ADCETRIS as a single agent was evaluated in a pivotal open-label, single-arm, multicenter study in 102 patients with relapsed or refractory HL. See Table 11 below for a summary of baseline patient and disease characteristics.
Table 11. Summary of baseline patient and disease characteristics in the phase 2 relapsed or refractory HL study:
| Patient characteristics | n=102 | ||
| Median age, years (range) | 31 years (15-77) | ||
| Gender | 48M (47%)/54F (53%) | ||
| ECOG status | |||
| 0 | 42 (41%) | ||
| 1 | 60 (59%) | ||
| Prior ASCT | 102 (100%) | ||
| Prior chemotherapy Regimens | 3.5 (1-13) | ||
| Time from ASCT to first post-transplant relapse | 6.7 mo (0-131) | ||
| Histologically confirmed CD30-expressing disease | 102 (100%) | ||
| Disease characteristics | |||
| Primary Refractory to frontline therapya | 72 (71%) | ||
| Refractory to most recent therapy | 43 (42%) | ||
| Baseline B symptoms | 35 (33%) | ||
| Stage III at initial diagnosis | 27 (26%) | Stage IV at initial diagnosis | 20 (20%) |
a Primary refractory HL is defined as a failure to achieve a complete remission to, or progressed within 3 months of completing frontline therapy.
Eighteen (18) patients (18%) received 16 cycles of ADCETRIS; and the median number of cycles received was 9 (ranging from 1 to 16).
Response to treatment with ADCETRIS was assessed by Independent Review Facility (IRF) using the Revised Response Criteria for Malignant Lymphoma (Cheson, 2007). Treatment response was assessed by spiral CT of chest, neck, abdomen and pelvis; PET scans and clinical data. Response assessments were performed at cycles 2, 4, 7, 10, 13, and 16 with PET at cycles 4 and 7.
The objective response rate (ORR) per IRF assessment was 75% (76 of 102 patients in the intent-to-treat [ITT] set) and tumour reduction was achieved in 94% of patients. Complete remission (CR) was 33% (34 of 102 patients in the ITT set). The median overall survival (OS) is 40.5 months (the median observation time (time to death or last contact) from first dose was 35.1 months (range 1.8 to 72.9+ months). The estimated overall survival rate at 5 years was 41% (95% CI [31%, 51%]). The investigator assessments were generally consistent with the independent review of the scans. Of the patients treated, 8 responding patients went on to receive an allogeneic SCT. For further efficacy results see Table 12.
Table 12. Efficacy results in relapsed or refractory Hodgkin lymphoma patients treated with 1.8 mg/kg of ADCETRIS every 3 weeks:
| Best clinical response (n=102) | IRF n (%) | 95% CI |
| Objective response rate (CR + PR) | 76 (75) | 64.9, 82.6 |
| Complete remission (CR) | 34 (33) | 24.3, 43.4 |
| Partial remission (PR) | 42 (41) | NA |
| Disease control rat (CR + PR + SD) | 98 (96) | 90.3, 98.9 |
| Duration of response | Median per IRF | 95% CI |
| Objective response rate (CR + PR)a | 6.7 months | 3.6, 14.8 |
| Complete remission (CR) | 27.9 months | 10.8, NEb |
| Overall survival | 95% CI | |
| Median | 40.5 months | 28.7, 61.9 |
| Estimated 5-year OS Rate | 41% | 31%, 51% |
a The range of DOR was 1.2+ months to 43+ months and the median follow-up time from first dose for patients who achieved objective response (OR) per IRF was 9.0 months.
b Not estimable.
An exploratory intra-patient analysis showed that approximately 64% of the HL patients treated with ADCETRIS as part of the SG035-0003 clinical study experienced an improvement in clinical benefit as measured by longer progression free survival (PFS) compared with their most recent prior line of therapy.
Of the 35 patients (33%) who had B symptoms at baseline, 27 patients (77%) experienced resolution of all B symptoms at a median time of 0.7 months from initiation of ADCETRIS.
Data in HL Patients Who Are Not Stem Cell Transplant (SCT) Candidates
Study-C25007
A phase 4 single-arm study was conducted in patients with relapsed or refractory HL (n=60) who had received at least one prior chemotherapeutic regimen and at the time of treatment initiation with ADCETRIS were not considered candidates for SCT or multiagent chemotherapy. Eligible patients were not to have received a prior SCT. The median number of cycles was 7 (range 1 to 16 cycles). Patients were treated with 1.8 mg/kg of ADCETRIS every 3 weeks.
At the time of the primary analysis of the primary endpoint, per IRF, the objective response rate (ORR) in the ITT population was 50% (95% CI, 37; 63%). A best overall response of CR was reported for 7 patients (12%); PR was reported for 23 patients (38%). Among these 30 patients, the median time to response, defined as the time from first dose to the soonest of PR or CR, was 6 weeks (range, 5 to 39 weeks). The median time to best overall response, defined as the time from first dose to the clinical best response of CR or PR, was 11 weeks (range, 5 to 60 weeks). Twenty-eight patients (47%) went on to receive SCT after a median of 7 cycles (range, 4 to 16 cycles) of ADCETRIS treatment. The 32 patients (53%) who did not receive subsequent SCT also received ADCETRIS for a median of 7 cycles (range, 1 to 16 cycles).
Of the 60 patients in the study, 49 patients (82%) received >1 prior cancer-related treatment and 11 patients (18%) received 1 prior cancer-related treatment. Per IRF, the ORR was 51% (95% CI [36%, 66%]) for the patients who had received >1 prior cancer-related treatment and 45% (95% CI [17%, 77%]) for the patients who had received 1 prior cancer-related treatment. For the patients who received >1 prior cancer-related treatment, a best overall response of CR was reported for 6 patients (12%); PR was reported for 19 patients (39%). For the patients who received 1 prior cancer-related treatment, CR was reported for 1 patient (9%) and PR was reported for 4 patients (36%). Out of the 49 patients receiving >1 line of prior treatment, 22 patients (45%) received subsequent SCT; of the 11 patients who had received 1 prior treatment, 6 patients (55%) received subsequent SCT.
Data were also collected from patients (n=15) in phase 1 dose escalation and clinical pharmacology studies, and from patients (n=26) in a NPP, with relapsed or refractory HL who had not received an ASCT, and who were treated with 1.8 mg/kg of ADCETRIS every 3 weeks.
Baseline patient characteristics showed failure from multiple prior chemotherapy regimens (median of 3 with a range of 1 to 7) before first administration with ADCETRIS. Fifty nine percent (59%) of patients had advanced stage disease (Stage III or IV) at initial diagnosis.
Results from these phase 1 studies and from the NPP experience showed, that in patients with relapsed or refractory HL without prior ASCT, clinically meaningful responses can be achieved as evidenced by an investigator-assessed, objective response rate of 54% and a complete remission rate of 22% after a median of 5 cycles of ADCETRIS.
Study SGN35-006 (Retreatment Study)
The efficacy of retreatment in patients who had previously responded (CR or PR) to treatment with ADCETRIS was evaluated in a phase 2, open-label, multicenter trial. Twenty patients with relapsed or refractory HL received a starting dose of 1.8 mg/kg and one patient received a starting dose of 1.2 mg/kg of ADCETRIS administered intravenously over 30 minutes every 3 weeks. The median number of cycles was 7 (range, 2 to 37 cycles). Of the 20 evaluable patients with HL, 6 patients (30%) achieved a CR and 6 patients (30%) achieved a PR with ADCETRIS retreatment, for an ORR of 60%. The median duration of response was 9.2 and 9.4 months in patients who achieved OR (CR+PR) and CR, respectively.
Systemic anaplastic large cell lymphoma
Study SGN35-014
The efficacy and safety of ADCETRIS were evaluated in a randomised, double-blind, double-dummy, active-controlled, multicenter trial of 452 patients with previously untreated CD30+ PTCL in combination with cyclophosphamide [C], doxorubicin [H] and prednisone [P] (CHP). For enrollment, the trial required CD30 expression ≥10% per immunohistochemistry. Only patients with CD30+ PTCLs who were eligible for a cyclophosphamide [C], doxorubicin [H], vincristine [O] and prednisone [P] (CHOP)-based regimen were included. The combination of ADCETRIS + CHP has not been studied in all PTCL subtypes. See Table 13 for enrolled PTCL subtypes. Of the 452 patients, 226 were randomised to treatment with ADCETRIS + CHP and 226 patients were randomised to treatment with CHOP. Randomisation was stratified by ALK-positive sALCL versus all other subtypes and by the International Prognostic Index (IPI) score. Patients were treated with 1.8 mg/kg of ADCETRIS administered as an intravenous infusion over 30 minutes on day 1 of each 21-day cycle + CHP (cyclophosphamide 750 mg/m² every 3 weeks by IV infusion; doxorubicin 50 mg/m² every 3 weeks by IV infusion; and prednisone 100 mg on Days 1 to 5 of each 3-week cycle, orally) for 6 to 29 8 cycles. The median number of cycles received was 6 (range, 1 to 8 cycles); 70% of patients received 6 cycles of treatment, and 18% received 8 cycles of treatment. Table 13 provides a summary of baseline patient and disease characteristics.
Table 13. Summary of baseline patient and disease characteristics in the phase 3 previously untreated PTCL study (ITT and sALCL):
| ITT Population | sALCL Populationb | |||
| Patient characteristics | ADCETRIS + CHP n=226 | CHOP n=226 | ADCETRIS + CHP n=162 | CHOP n=154 |
| Median age (range) | 58.0 (18-85) | 58.0 (18-83) | 55.0 (18-85) | 54.0 (18-83) |
| Patients ≥65 years old (%) | 69 (31) | 70 (31) | 38 (23) | 36 (23) |
| Male sex, n (%) | 133 (59) | 151 (67) | 95 (59) | 110 (71) |
| ECOG status, n (%) | ||||
| 0 | 84 (37) | 93 (41) | 58 (36) | 53 (34) |
| 1 | 90 (40) | 86 (38) | 62 (38) | 61 (40) |
| 2 | 51 (23) | 47 (21) | 41 (25) | 40 (26) |
| Disease characteristics | ||||
| Diagnosis, per local assessment, n (%)a | ||||
| sALCL | 162 (72) | 154 (68) | 162 (100) | 154 (100) |
| ALK-positive | 49 (22) | 49 (22) | 49 (30) | 49 (32) |
| ALK-negative | 113 (50) | 105 (46) | 113 (70) | 105 (68) |
| Peripheral T-cell lymphoma (PTCL-NOS) | 29 (13) | 43 (19) | NA | NA |
| Angioimmunoblastic T-cell lymphoma (AITL) | 30 (13) | 24 (11) | NA | NA |
| Adult T-cell leukemia/lymphoma (ATLL) | 4 (2) | 3 (1) | NA | NA |
| Enteropathy-associated T-cell lymphoma (EATL) | 1 (0) | 2 (1) | NA | NA |
| Median time from diagnosis to first dose, months (range) | 0.8 (0, 19) | 0.9 (0, 10) | 0.8 (0, 19) | 0.9 (0, 10) |
| Disease stage at initial diagnosis of PTCL, n (%) | ||||
| Stage I | 12 (5) | 9 (4) | 12 (7) | 7 (5) |
| Stage II | 30 (13) | 37 (16) | 22 (14) | 27 (18) |
| Stage III | 57 (25) | 67 (30) | 29 (18) | 46 (30) |
| Stage IV | 127 (56) | 113 (50) | 99 (61) | 74 (48) |
| IPI score | ||||
| 0 | 8 (4) | 16 (7) | 7 (4) | 14 (9) |
| 1 | 45 (20) | 32 (14) | 34 (21) | 18 (12) |
| 2 | 74 (33) | 78 (35) | 58 (36) | 60 (39) |
| 3 | 66 (29) | 66 (29) | 37 (23) | 40 (26) |
| 4 | 29 (13) | 25 (11) | 22 (14) | 16 (10) |
| 5 | 4 (2) | 9 (4) | 4 (2) | 6 (4) |
| Extranodal involvement at time of diagnosis, n (%) | ||||
| ≤1 site | 142 (63) | 146 (65) | 94 (58) | 95 (62) |
| >1 site | 84 (37) | 80 (35) | 68 (42) | 59 (38) |
| Baseline bone marrow biopsy- lymphoma involvement, n (%) | ||||
| Yes | 30 (13) | 34 (15) | 15 (9) | 13 (8) |
| No | 196 (87) | 192 (85) | 147 (91) | 141 (92) |
The primary endpoint in SGN35-014 was PFS per IRF, defined as the time from the date of randomisation to the date of first documentation of progressive disease, death due to any cause, or receipt of subsequent anticancer chemotherapy to treat residual or progressive disease, whichever occurs first. Receipt of post-treatment consolidative radiotherapy, post-treatment chemotherapy for the purpose of mobilising peripheral blood stem cells, or consolidative autologous or allogeneic stem cell transplant were not considered as disease progression or as having started new anticancer therapy.
Key secondary endpoints included PFS per IRF for patients with centrally-confirmed sALCL, CR rate per IRF following the completion of study treatment, OS and ORR per IRF following the completion of study treatment which were tested by a fixed sequence testing procedure following the statistical significance of PFS per IRF.
The primary endpoint and alpha-protected, key secondary endpoints, which were evaluated hierarchically, were met. The median PFS per IRF for the ITT population was 48.2 months on the ADCETRIS + CHP arm versus 20.8 months on the CHOP arm. The stratified hazard ratio was 0.71 (95% CI: 0.54; 0.93, p=0.011), indicating a 29% reduction in the risk of PFS events for ADCETRIS + CHP versus CHOP. For overall survival, the stratified hazard ratio was 0.66 (95% CI: 0.46; 0.95, p=0.024), a 34% reduction in the risk of OS events for ADCETRIS + CHP versus CHOP.
PFS per IRF for patients with centrally-confirmed sALCL was a pre-specified key secondary endpoint. The median PFS per IRF was 55.7 months on the ADCETRIS + CHP arm versus 54.2 months on the CHOP arm. The stratified hazard ratio was 0.59 (95% CI: 0.42; 0.84), compatible with a statistically significant 41% reduction in the risk of PFS events for ADCETRIS + CHP versus CHOP (p-value=0.003), see Figure 6 and Table 14.
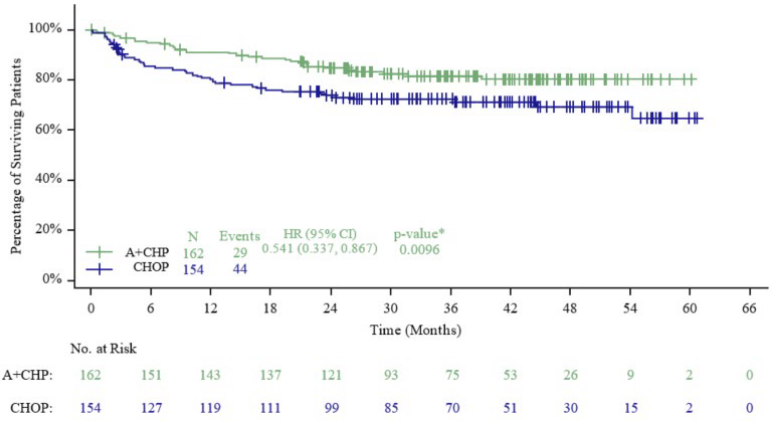
Subgroup analyses were performed for patients with locally-diagnosed sALCL. For overall survival, the stratified hazard ratio was 0.54 (95% CI: 0.34; 0.87), a 46% reduction in the risk of OS events for ADCETRIS + CHP versus CHOP, see Figure 7. At the end of treatment, the CR rate by IRF assessment was 71.0% for patients on the ADCETRIS + CHP arm compared with 53.2% for patients on the CHOP arm with a difference of 17.7% (95% CI: 7.2%; 28.3%). At the end of treatment, the ORR rate by IRF assessment was 87.7% for patients on the ADCETRIS + CHP arm compared with 70.8% for patients on the CHOP arm with a difference of 16.9% (95% CI: 8.1%; 25.7%). In the subgroup of patients with ALK+ sALCL and ALK- sALCL the stratified hazard ratio for PFS per IRF was 0.29 (95% CI: 0.11; 0.79) and 0.65 (95% CI: 0.44; 0.95), respectively.
Table 14. Efficacy results in patients with previously untreated sALCL with 1.8 mg/kg of ADCETRIS on day 1 of a 3-week cycle (primary analysis):
| ADCETRIS + CHP n=162a | CHOP n=154a | ||||
| PFS per IRF | Number of patients with a PFS event, n (%) | 56 (34) | 73 (48) | ||
| Median PFS, months (95% CI) | 55.66 (48.20, NE) | 54.18 (13.44, NE) | |||
| Hazard ratio (95%b | 0.59 (0.42, 0.84) | ||||
| p-valuec | 0.0031 | ||||
| Estimated PFS (95% CI)d at: | |||||
| 6 months | 88.0% (81.8%, 92.2%) | 68.4% (60.3%, 75.2%) | |||
| 12 months | 78.7% (71.4%, 84.4%) | 60.3% (51.9%, 67.6%) | |||
| 24 months | 68.4% (60.4%, 75.2%) | 53.9% (45.5%, 61.5%) | |||
| 36 months | 65.5% (57.1%, 72.7%) | 50.2% (41.6%, 58.1%) | |||
| OSe | |||||
| Number of deaths (%) | 29 (18) | 44 (29) | |||
| Median OS, months (95% CI) | NE (NE, NE) | NE (NE, NE) | |||
| Hazard ratio (95% CI)b | 0.54 (0.34, 0.87) | ||||
| p-valuec,f | 0.0096 | ||||
| CR Rateg | |||||
| % (95% CI) | 71% (63.3, 77.8) | 53% (45.0, 61.3) | |||
| p-valuef,h | 0.0004 | ||||
| ORRg | |||||
| % (95% CI) | 88% (81.6, 92.3) | 71% (62.9, 77.8) | |||
| p-valuef,h | <0.0001 | ||||
CR = complete remission; IRF = Independent Review Facility; NE: Not estimable; ORR = objective response rate; PFS = progression-free survival.
a PFS per IRF is calculated using patients with centrally-confirmed sALCL, with n=163 patients in A+CHP arm and n=151 in CHOP arm. OS, CR, and ORR are calculated using patients with locally-diagnosed sALCL
b Hazard ratio (A+CHP/CHOP) and 95% confidence intervals are based on a stratified Cox's proportional hazard regression model with stratification factors (ALK-positive sALCL versus all others and International Prognostic Index [IPI] score at baseline). Hazard ratio <1 favours A+CHP arm.
c p-value is calculated using a stratified log-rank test.
d PFS rate is estimated using Kaplan-Meier methods and 95% CI is calculated using the complementary log-log transformation method.
e Median OS follow-up in the A+CHP arm was 38.5 months; in the CHOP arm was 41.0 months.
f p-value is not adjusted for multiplicity.
g Response per 2007 International Working Group Criteria at end of treatment.
h p-value is calculated using a stratified Cochran-Mantel-Haenszel test.
Figure 6. Progression-free survival per IRF in the sALCL population (ADCETRIS + CHP vs. CHOP) (primary analysis):
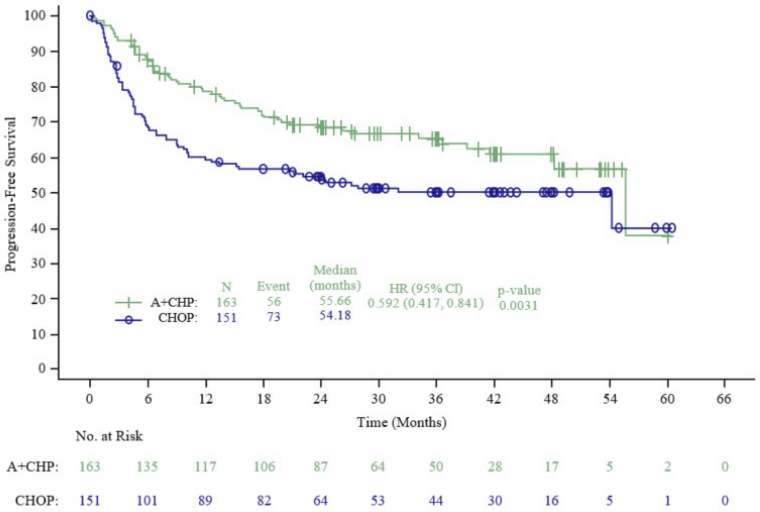
Figure 7. Overall survival in the sALCL population (ADCETRIS + CHP vs. CHOP) (primary analysis):
* p-value for overall survival is not adjusted for multiplicity.
As of study closure more than 7 years after enrollment of the first patient, PFS per investigator results in the ITT population indicated a 30% reduction in the risk of a PFS event in the ADCETRIS+CHP arm compared with patients treated with CHOP (HR=0.70 [95% CI (0.53, 0.91)]). PFS per investigator results in the sALCL population indicated a 45% reduction in the risk of a PFS event in the ADCETRIS+CHP arm compared with patients treated with CHOP (HR=0.55 [95% CI (0.39, 0.79)]).
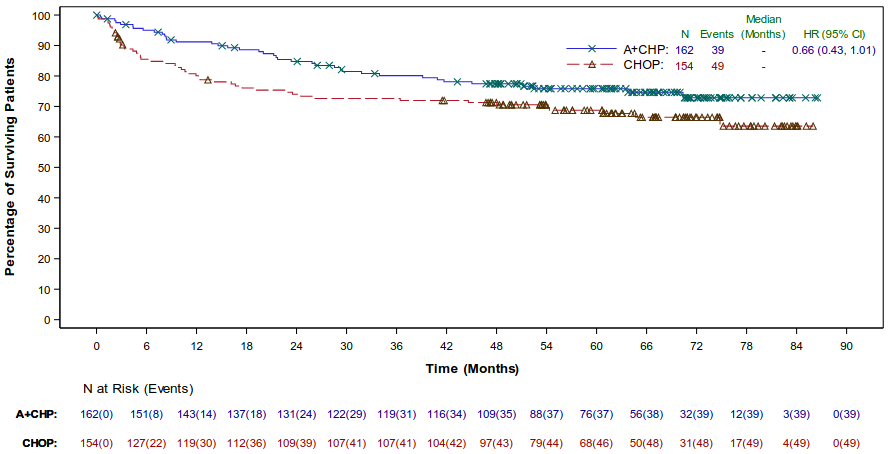
As of study closure, overall survival results continued to show a benefit and were consistent with those reported at the time of the primary analysis. Overall survival results in the ITT population indicated a 28% reduction in the risk of death in the ADCETRIS+CHP arm compared with patients treated with CHOP (HR=0.72 [95% CI (0.53 to 0.99)]). Overall survival results in the sALCL population indicated a 34% reduction in the risk of death in the ADCETRIS+CHP arm compared with patients treated with CHOP (HR=0.66 [95% CI (0.43, 1.01)]), see Figure 8.
Figure 8. Overall survival in the sALCL population (ADCETRIS + CHP vs. CHOP) (study closure):
Study SG035-0004
The efficacy and safety of ADCETRIS as a single agent was evaluated in an open-label, single-arm, multicenter study in 58 patients with relapsed or refractory sALCL. See Table 15 below for a summary of baseline patient and disease characteristics.
Table 15. Summary of baseline patient and disease characteristics in the phase 2 relapsed or refractory sALCL study:
| Patient characteristics | n=58 |
| Median age, years (range) | 52 years (14-76) |
| Gender | 33M (57%)/25F (43%) |
| ECOG statusa | |
| 0 | 19 (33%) |
| 1 | 38 (66%) |
| Prior ASCT | 15 (26%) |
| Prior chemotherapy Regimens (range) | 2 (1-6) |
| Histologically confirmed CD30-expressing disease | 57 (98%) |
| Anaplastic lymphoma kinase (ALK)-negative disease | 42 (72%) |
| Disease characteristics | |
| Primary Refractory to frontline therapyb | 36 (62%) |
| Refractory to most recent therapy | 29 (50%) |
| Relapsed to most recent therapy | 29 (50%) |
| Baseline B symptoms | 17 (29%) |
| Stage III at initial diagnosis | 8 (14%) |
| Stage IV at initial diagnosis | 21 (36%) |
a One patient had a baseline ECOG status of 2, which was prohibited by protocol and is captured as Inclusion Criteria Not Met.
b Primary refractory sALCL is defined as a failure to achieve a complete remission to, or progressed within 3 months of completing frontline therapy.
The median time from initial sALCL diagnosis to first dose with ADCETRIS was 16.8 months.
Ten (10) patients (17%) received 16 cycles of ADCETRIS; the median number of cycles received was 7 (range, 1 to 16).
Response to treatment with ADCETRIS was assessed by Independent Review Facility (IRF) using the Revised Response Criteria for Malignant Lymphoma (Cheson, 2007). Treatment response was assessed by spiral CT of chest, neck, abdomen and pelvis; PET scans and clinical data. Response assessments were performed at cycles 2, 4, 7, 10, 13 and 16 with PET at cycles 4 and 7.
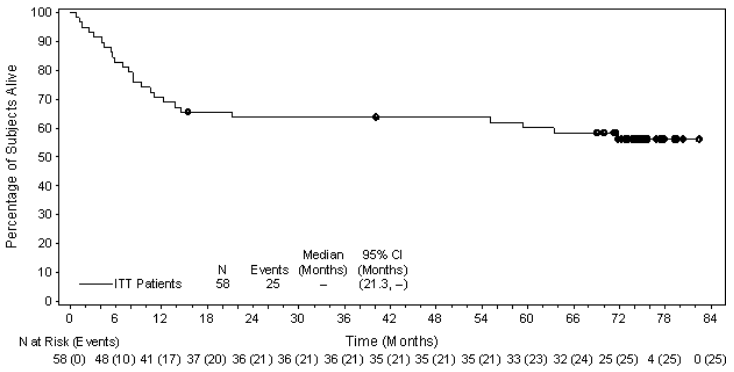
The ORR per IRF assessment was 86% (50 of 58 patients in the ITT set). CR was 59% (34 of 58 patients in the ITT set) and tumour reduction (of any degree) was achieved in 97% of patients. The estimated overall survival at 5 years was 60% (95% CI [47%,73%]). The median observation time (time to death or last contact) from first dose was 71.4 months. The investigator assessments were generally consistent with the independent review of the scans. Of the patients treated, 9 responding patients went on to receive an allogeneic stem cell transplant (SCT) and 9 responding patients went on to autologous SCT. For further efficacy results, see Table 16 and Figure 9.
Table 16. Efficacy results in relapsed or refractory sALCL patients treated with 1.8 mg/kg of ADCETRIS every 3 weeks:
| Best clinical response (n=58) | IRF n (%) | 95% CI |
| Objective response rate (CR + PR) | 50 (86) | 74.6, 93.9 |
| Complete remission (CR) | 34 (59) | 44.9, 71.4 |
| Partial remission (PR) | 16 (28) | NA |
| Disease control rat (CR + PR + SD) | 52 (90) | 78.8, 96.1 |
| Duration of response | Median per IRF | 95% CI |
| Objective response (CR + PR)a | 13.2 | 5.7, 26.3 |
| Complete remission (CR) | 26.3 | 13.2, NEb |
| Progression Free Survival | Median per IRF | 95% CI |
| Median | 14.6 | 6.9, 20.6 |
| Overall survival | Median | 95% CI |
| Median | Not reached | 21.3, NEb |
Figure 9. Kaplan-Meier Plot of OS:
An exploratory intra-patient analysis showed that approximately 69% of the sALCL patients treated with ADCETRIS as part of the SG035-0004 clinical study experienced an improvement in clinical benefit as measured by longer progression free survival (PFS) compared with their most recent prior line of therapy.
Of the 17 patients (29%) who had B symptoms at baseline, 14 patients (82%) experienced resolution of all B symptoms in a median time from initiation of ADCETRIS of 0.7 months.
Study C25006
The efficacy and safety of ADCETRIS as a single agent were also evaluated in a phase 4 open-label, single-arm multicenter study in 50 patients with relapsed or refractory sALCL. The ORR per IRF assessment was 64% (32 of 50 patients in the ITT set). The median DOR per IRF was not reached (95% CI 19.71 months, NE). The CR rate was 30% (15 of 50 patients in the ITT set), and tumour reduction (of any degree) was achieved in 93% of evaluable patients. The median DOCR per IRF was not reached (95% CI 10.61 months, NE). Response assessments were generally consistent between IRF and investigator. Of the patients treated, 13 patients went on to receive a haematopoietic stem cell transplant.
Pooled data from studies C25006 and SG035-0004 (N=108) show an ORR per IRF of 76% (82 of 108 patients in the ITT set). The median DOR per IRF was 17.0 months (95% CI 12.62, 32.46). CR was 45% (49 of 108 patients in the ITT set) and tumour reduction (of any degree) was achieved in 96% of evaluable patients. The median DOCR per IRF was 26.3 months (95% CI 16.16, NE). Response assessments per IRF and investigator were generally consistent.
Study SGN35-006 (Retreatment study)
The efficacy of retreatment in patients who had previously responded (CR or PR) to treatment with ADCETRIS was evaluated in a phase 2, open-label, multicenter trial. Seven patients with relapsed sALCL received a starting dose of 1.8 mg/kg and one patient received a starting dose of 1.2 mg/kg of ADCETRIS administered intravenously over 30 minutes every 3 weeks. The median number of cycles was 8.5 (range, 2 to 30 cycles). Of the 8 sALCL patients, 3 were retreated twice for a total of 11 retreatment experiences. Retreatment with ADCETRIS resulted in 6 CRs (55%) and 4 PRs (36%), for an ORR of 91%. The median duration of response was 8.8 and 12.3 months in patients who achieved OR (CR+PR) and CR, respectively.
Cutaneous T-cell lymphoma
Study C25001
The efficacy and safety of ADCETRIS as a single agent was evaluated in a pivotal phase 3, open-label, randomised, multicentre study in 128 patients with histologically confirmed CD30+ CTCL. CD30 positivity was defined as ≥10% target lymphoid cells demonstrating membrane, cytoplasmic, and/or Golgi staining pattern based on an immunohistochemistry assay (Ventana anti-CD30 [Ber-H2]). Patients with a diagnosis of mycosis fungoides [MF] or primary cutaneous anaplastic large cell lymphoma [pcALCL] were considered eligible for the study. Patients were stratified by these disease types and randomised 1:1 to receive either ADCETRIS or the physician's choice of either methotrexate or bexarotene. Patients with pcALCL received either prior radiation therapy or at least 1 prior systemic therapy and patients with MF received at least 1 prior systemic therapy. Patients with a concurrent diagnosis of systemic ALCL, Sezary syndrome and other non-Hodgkin lymphoma (except for lymphomatoid papulosis [LyP]) were excluded from this study. Patients were treated with 1.8 mg/kg of ADCETRIS intravenously over 30 minutes every 3 weeks for up to 16 cycles or physician's choice for up to 48 weeks. The median number of cycles was approximately 12 cycles in the ADCETRIS arm. In the physician's choice arm, the median duration of treatment (number of cycles) for patients receiving bexarotene was approximately 16 weeks (5.5 cycles) and 11 weeks (3 cycles) for patients receiving methotrexate. Table 17 provides a summary of the baseline patient and disease characteristics.
Table 17. Summary of baseline patient and disease characteristics in the phase 3 CTCL Study (ITT Population):
| Patient characteristics | ADCETRIS n=64 | Physician's Choice (Methotrexate or Bexarotene) n=64 |
| Median age (range) | 62 years (22-83) | 58.5 years (22-83) |
| Patients ≥65 years old n (%) | 28 (44%) | 24 (38%) |
| Gender n (%) | 33M (52%)/ 31F (48%) | 37M (58%)/ 27F (42%) |
| ECOG status n (%) | ||
| 0 | 43 (67) | 46 (72) |
| 1 | 18 (28) | 16 (25) |
| 2 | 3 (5) | 2 (3) |
| Disease characteristics | ||
| Median number of prior therapies (range) | 4 (0-13) | 3.5 (1-15) |
| Median number of skin-directed therapies (range) | 1 (0-6) | 1 (0-9) |
| Median number of systemic therapies (range) | 2 (0-11) | 2 (1-8) |
| MF, n (%) | 48 (75) | 49 (77) |
| Early (IA-IIA) | 15 (31) | 18 (37) |
| Advanced (IIB-IVBa) | 32 (67) | 30 (61) |
| pcALCL, n (%) | 16 (25) | 15 (23) |
| Skin only | 9 (56) | 11 (73) |
| Extracutaneous disease | 7 (44) | 4 (27) |
a One patient in each arm had incomplete staging data and are not included in the table.
The most common prior skin directed therapies in the ITT population were radiotherapy (64%), phototherapy (48%) and topical steroids (17%). The most common prior systemic therapies in the ITT population were chemotherapy (71%), immunotherapy (43%) and bexarotene (38%).
The primary endpoint was objective response rate that lasts at least 4 months (ORR4) (duration from first response to last response ≥4 months), as determined by an independent review of the Global Response Score (GRS) consisting of skin evaluations (modified severity weighted assessment tool [mSWAT] as assessed per investigator), nodal and visceral radiographic assessment, and detection of circulating Sézary cells (Olsen 2011). Table 18 includes the results for ORR4 and other key secondary endpoints.
Table 18. Efficacy results in CTCL patients treated with 1.8 mg/kg of ADCETRIS every 3 weeks (ITT population):
| ADCETRIS (n=64) | Physician's Choice (Methotrexate or Bexarotene) n=64 | |
| Objective Response Rate lasting at least 4 months (ORR4) per IRF | ||
| n (%) | 36 (56,3) | 8 (12,5) |
| Percent Difference (95% CI) | 43.8 (29.1, 58.4) | |
| p-value | <0.001 | |
| Complete Response (CR) per IRF | ||
| n (%) | 10 (15.6) | 1 (1.6) |
| Percent Difference (95% CI) | 14.1 (-4.0, 31.5) | |
| Adjusted p-valuea | 0.0046 | |
| Progression Free Survival (PFS) per IRF | ||
| Median (months) | 16.7 | 3.5 |
| Hazard Ratio | 0.270 | |
| 95% CI | (0.17, 0.43) | |
| Adjusted p-valuea | <0.001 | |
a Calculated from a weighted Holm's procedure.
Pre-specified subgroup analyses of ORR4 per IRF were performed by patients' CTCL subtype, physicians' choice of treatment, baseline ECOG status, age, gender, and geographic region. The analyses showed a consistent trend towards benefit for patients who received ADCETRIS compared with patients who received physician's choice. ORR4 was 50% and 75% in the ADCETRIS arm versus 10.2% and 20% in the physician's choice arm for MF and pcALCL, respectively.
No meaningful differences in quality of life (assessed by the EuroQol five dimensions questionnaire [EQ-5D] and Functional Assessment of Cancer Therapy-General [FACT-G]) were observed between the treatment arms.
The efficacy and safety of ADCETRIS were evaluated in two additional open-label studies in 108 patients with relapsed CD30+ CTCL (including MF and pcALCL as well as SS, LyP and mixed CTCL histology), regardless of CD30 expression level. Patients were treated with ADCETRIS 1.8 mg/kg intravenously over 30 minutes every 3 weeks for up to 16 cycles. The safety and efficacy results in these studies were consistent with results in Study C25001. Overall response rates for MF were 54-66%; pcALCL, 67%; SS, 50%; LyP, 92%; and mixed CTCL histology, 82-85%.
Paediatric population
Combination therapy
C25004
The safety and anti-tumour activity of ADCETRIS were evaluated in an open-label, multicenter trial in 59 paediatric patients (6-17 years of age) with previously untreated advanced-stage classical CD30+ HL in combination with chemotherapy (doxorubicin [A], vinblastine [V] and dacarbazine [D] [AVD]). All patients had a histologically confirmed CD30-expressing disease. Fifty-nine percent of patients (n=35) had extranodal site involvement. All 59 paediatric patients were treated on days 1 and 15 of each 28-day cycle with 48 mg/m² of ADCETRIS administered as an intravenous infusion over 30 minutes + doxorubicin 25 mg/m², vinblastine 6 mg/m², and dacarbazine 375 mg/m². The BSA-based dose of ADCETRIS was chosen to match the observed PK exposures in adults in Study C25003. The paediatric maximum tolerated dose (MTD) was not reached. The majority of patients (88%) achieved an objective response by IRF assessment at the EOT, with 76% achieving a CR. No patient died. A total of 13 patients (22%) in the safety population were reported to have received irradiation after Cycle 6.
Monotherapy
C25002
The safety, pharmacokinetics and anti-tumour activity of ADCETRIS in 36 paediatric patients (7-17 years of age) with r/r HL and sALCL (children aged 7-11 years, n=12 and adolescents aged 12 to 17 years, n=24) were evaluated in a phase ½ open-label, single-agent, multicentre dose-escalation study (C25002). Phase 1 of the study assessed the safety profile (see section 4.8), determined the paediatric maximum tolerated dose (MTD) and/or recommended phase 2 dose (RP2D), and assessed the pharmacokinetics of ADCETRIS (see section 5.2). Phase 1 included 3 r/r HL patients treated at 1.4 mg/kg and 9 patients (7 r/r HL and 2 sALCL) treated at 1.8 mg/kg. The MTD was not reached. The RP2D was determined to be 1.8 mg/kg. Across the study, a total of 16 patients with r/r HL and 17 patients with r/r sALCL, of whom 10 were in first relapse, were treated with 1.8 mg/kg of ADCETRIS. The overall response rate (ORR) per independent review facility (IRF) was analysed across both study phases at the RP2D. Of these 33 patients who received the RP2D, 32 were evaluable for response. The ORR was 47% in response-evaluable patients with r/r HL, 53% in patients with r/r sALCL and 60% in sALCL patients in first relapse. Eight HL patients and 9 sALCL patients went on to receive SCT following treatment with ADCETRIS.
Pharmacokinetic properties
Monotherapy
The pharmacokinetics of brentuximab vedotin were evaluated in phase 1 studies and in a population pharmacokinetic analysis of data from 314 patients. In all clinical trials, brentuximab vedotin was administered as an intravenous infusion.
Maximum concentrations of brentuximab vedotin ADC were typically observed at the end of infusion or the sampling timepoint closest to the end of infusion. A multiexponential decline in ADC serum concentrations was observed with a terminal half-life of approximately 4 to 6 days. Exposures were approximately dose proportional. Minimal to no accumulation of ADC was observed with multiple doses at the every 3-week schedule, consistent with the terminal half-life estimate. Typical Cmax and AUC of ADC after a single 1.8 mg/kg in a phase 1 study was approximately 31.98 μg/mL and 79.41 μg/mL x day respectively.
MMAE is the major metabolite of brentuximab vedotin. Median Cmax, AUC and Tmax of MMAE after a single 1.8 mg/kg of the ADC in a phase 1 study was approximately 4.97 ng/mL, 37.03 ng/mL x day and 2.09 days respectively. MMAE exposures decreased after multiple doses of brentuximab vedotin with approximately 50% to 80% of the exposure of the first dose being observed at subsequent doses. MMAE is further metabolised mainly to an equally potent metabolite; however, its exposure is an order of magnitude lower than that of MMAE. Thus, it is not likely to have any substantial contribution to the systemic effects of MMAE.
In the first cycle, higher MMAE exposure was associated with an absolute decrease in neutrophil count.
Combination therapy
The pharmacokinetics of ADCETRIS in combination with AVD were evaluated in a single phase 3 study in 661 patients. Population pharmacokinetic analysis indicated that the pharmacokinetics of ADCETRIS in combination with AVD were consistent to that in monotherapy.
After multiple-dose, IV infusion of 1.2 mg/kg brentuximab vedotin every two weeks, maximal serum concentrations of ADC were observed near the end of the infusion and elimination exhibited a multi-exponential decline with a t1/2z of approximately 4 to 5 days. Maximal plasma concentrations of MMAE were observed approximately 2 days after the end of infusion, and exhibited a mono-exponential decline with a t1/2z of approximately 3 to 4 days.
After multiple-dose, IV infusion of 1.2 mg/kg brentuximab vedotin every two weeks, steady-state trough concentrations of ADC and MMAE were achieved by Cycle 3. Once steady-state was achieved, the PK of ADC did not appear to change with time. ADC accumulation (as assessed by AUC14D between Cycle 1 and Cycle 3) was 1.27-fold. The exposure of MMAE (as assessed by AUC14D between Cycle 1 and Cycle 3) appeared to decrease with time by approximately 50%.
The pharmacokinetics of ADCETRIS in combination with CHP were evaluated in a single phase 3 study in 223 patients (SGN35-014). After multiple-dose IV infusion of 1.8 mg/kg ADCETRIS every 3 weeks, the pharmacokinetics of ADC and MMAE were similar to those of monotherapy.
Distribution
In vitro, the binding of MMAE to human serum plasma proteins ranged from 68-82%. MMAE is not likely to displace or to be displaced by highly protein-bound medicines. In vitro, MMAE was a substrate of P-gp and was not an inhibitor of P-gp at clinical concentrations.
In humans, the mean steady state volume of distribution was approximately 6-10 L for ADC. Based on population PK estimation the typical apparent central volume of distribution of MMAE was 35.5 L.
Metabolism
The ADC is expected to be catabolised as a protein with component amino acids recycled or eliminated.
In vivo data in animals and humans suggest that only a small fraction of MMAE released from brentuximab vedotin is metabolised. The levels of MMAE metabolites have not been measured in human plasma. At least one metabolite of MMAE has been shown to be active in vitro.
MMAE is a substrate of CYP3A4 and possibly CYP2D6. In vitro data indicate that the MMAE metabolism that occurs is primarily via oxidation by CYP3A4/5. In vitro studies using human liver microsomes indicate that MMAE inhibits only CYP3A4/5 at concentrations much higher than was achieved during clinical application. MMAE does not inhibit other isoforms.
MMAE did not induce any major CYP450 enzymes in primary cultures of human hepatocytes.
Elimination
The ADC is eliminated by catabolism with a typical estimated CL and half-life of 1.5 L/day and 4-6 days respectively.
The elimination of MMAE was limited by its rate of release from ADC, typical apparent CL and half-life of MMAE was 19.99 L/day and 3-4 days respectively.
An excretion study was undertaken in patients who received a dose of 1.8 mg/kg of brentuximab vedotin. Approximately 24% of the total MMAE administered as part of the ADC during a brentuximab vedotin infusion was recovered in both urine and faeces over a 1-week period. Of the recovered MMAE, approximately 72% was recovered in the faeces. A lesser amount of MMAE (28%) was excreted in the urine.
Pharmacokinetics in special populations
Population PK analysis showed that baseline serum albumin concentration was a significant covariate of MMAE clearance. The analysis indicated that MMAE clearance was 2-fold lower in patients with low serum albumin concentrations < 3.0 g/dL compared with patients with serum albumin concentrations within the normal range.
Hepatic impairment
A study evaluated the PK of brentuximab vedotin and MMAE after the administration of 1.2 mg/kg of ADCETRIS to patients with mild (Child-Pugh A; n=1), moderate (Child-Pugh B; n=5) and severe (Child-Pugh C; n=1) hepatic impairment. Compared to patients with normal hepatic function, MMAE exposure increased approximately 2.3-fold (90% CI 1.27-4.12-fold) in patients with hepatic impairment.
Renal impairment
A study evaluated the PK of brentuximab vedotin and MMAE after the administration of 1.2 mg/kg of ADCETRIS to patients with mild (n=4), moderate (n=3) and severe (n=3) renal impairment. Compared to patients with normal renal function, MMAE exposure increased approximately 1.9-fold (90% CI 0.85-4.21-fold) in patients with severe renal impairment (creatinine clearance <30 mL/min). No effect was observed in patients with mild or moderate renal impairment.
Elderly
The population pharmacokinetics of brentuximab vedotin were examined from several studies, including data from 380 patients up to 87 years old (34 patients ≥65 - <75 and 17 patients ≥75 years of age). Additionally, the population pharmacokinetics of brentuximab vedotin in combination with AVD were examined, including data from 661 patients up to 82 years old (42 patients ≥65 - <75 and 17 patients ≥75 years of age). The influence of age on pharmacokinetics was investigated in each analysis and it was not a significant covariate.
Paediatric population
Monotherapy:
C25002
The pharmacokinetics of brentuximab vedotin ADC and MMAE following a 30-minute intravenous infusion of BV administered at 1.4 mg/kg or 1.8 mg/kg given every 3 weeks were evaluated in a phase ½ clinical trial of 36 paediatric patients (7-17 years of age) with r/r HL and sALCL (children aged 7-11 years, n=12 and adolescents aged 12 to 17 years, n=24) (see section 5.1). The Cmax of ADC was typically observed at the end of infusion or the sampling closest to the end of infusion. A multi-exponential decline in ADC serum concentrations was observed with a terminal half-life of approximately 4 to 5 days. Exposures were approximately dose proportional with a trend observed for lower ADC exposures at lower ages/body weights in the study population.
Median ADC AUC in children and adolescents from this study was approx. 14% and 3% lower than in adult patients, respectively, while MMAE exposures were 53% lower and 13% higher, respectively, than in adult patients. Median Cmax and AUC of ADC after a single 1.8 mg/kg dose were 29.8 μg/mL and 67.9 μg*day/mL, respectively, in patients <12 years of age and 34.4 μg/mL and 77.8 μg*day/mL, respectively, in patients ≥12 years of age. Median Cmax, AUC, and Tmax of MMAE after a single 1.8 mg/kg dose were 3.73 ng/mL, 17.3 ng*day/mL, and 1.92 days, respectively, in patients <12 years of age and 6.33 ng/mL, 42.3 ng*day/mL, and 1.82 days, respectively, in patients ≥12 years of age. There was a trend of increased clearance of brentuximab vedotin in paediatric patients confirmed positive for ADAs. No patients aged <12 years (0 of 11) and 2 patients aged ≥12 years (2 of 23) became persistently ADA positive.
Combination therapy:
C25004
The pharmacokinetics of brentuximab vedotin ADC and MMAE following a 30-minute intravenous infusion of BV administered at 48 mg/m² every 2 weeks in combination with doxorubicin, vinblastine, and dacarbazine (AVD) were evaluated in a phase ½ clinical trial of 59 paediatric patients (6-17 years of age) with advanced-stage newly diagnosed CD30+ classical Hodgkin lymphoma (children aged 6-11 years, n=11 and adolescents aged 12 to 17 years, n=48). The Cmax of ADC occurred in serum approximately at the end of infusion and declined in a multiexponential manner with a terminal half-life of approximately 4 days. The Cmax of MMAE occurred in plasma approximately 2 days following BV administration with a half-life of approximately 2 days. Geometric mean Cmax and AUC of ADC following a single 48 mg/m² dose were 22.5 μg/mL and 46.7 μg*day/mL, respectively. Geometric mean Cmax and AUC of MMAE following a single 48 mg/m² dose were 4.9 ng/mL and 27.2 ng*day/mL, respectively. Similar ADC exposures were achieved following body surface area-based dosing of BV at 48 mg/m² in combination with AVD among paediatric age groups (<12 years, 12–16 years and >16 years).
Preclinical safety data
MMAE has been shown to have aneugenic properties in an in vivo rat bone marrow micronucleus study. These results were consistent with the pharmacological effect of MMAE on the mitotic apparatus (disruption of the microtubule network) in cells.
The effects of brentuximab vedotin on human male and female fertility have not been studied. However, results of repeat-dose toxicity studies in rats indicate the potential for brentuximab vedotin to impair male reproductive function and fertility. Testicular atrophy and degeneration were partially reversible following a 16-week treatment-free period.
Brentuximab vedotin caused embryo-foetal lethality in pregnant female rats.
In nonclinical studies, lymphoid depletion and reduced thymic weight were observed, consistent with the pharmacologic disruption of microtubules caused by MMAE derived from brentuximab vedotin.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.