AFINITOR Tablet Ref.[50349] Active ingredients: Everolimus
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors
ATC code: L01EG02
Mechanism of action
Everolimus is a selective mTOR (mammalian target of rapamycin) inhibitor. mTOR is a key serine-threonine kinase, the activity of which is known to be upregulated in a number of human cancers. Everolimus binds to the intracellular protein FKBP-12, forming a complex that inhibits mTOR complex-1 (mTORC1) activity. Inhibition of the mTORC1 signalling pathway interferes with the translation and synthesis of proteins by reducing the activity of S6 ribosomal protein kinase (S6K1) and eukaryotic elongation factor 4E-binding protein (4EBP-1) that regulate proteins involved in the cell cycle, angiogenesis and glycolysis. S6K1is thought to phosphorylate the activation function domain 1 of the oestrogen receptor, which is responsible for ligand-independent receptor activation. Everolimus reduces levels of vascular endothelial growth factor (VEGF), which potentiates tumour angiogenic processes. Everolimus is a potent inhibitor of the growth and proliferation of tumour cells, endothelial cells, fibroblasts and blood-vessel-associated smooth muscle cells and has been shown to reduce glycolysis in solid tumours in vitro and in vivo.
Clinical efficacy and safety
Hormone receptor-positive advanced breast cancer
BOLERO-2 (study CRAD001Y2301), a randomised, double-blind, multicentre phase III study of Afinitor + exemestane versus placebo + exemestane, was conducted in postmenopausal women with oestrogen receptor-positive, HER2/neu negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole. Randomisation was stratified by documented sensitivity to prior hormonal therapy and by the presence of visceral metastasis. Sensitivity to prior hormonal therapy was defined as either (1) documented clinical benefit (complete response [CR], partial response [PR], stable disease ≥24 weeks) from at least one prior hormonal therapy in the advanced setting or (2) at least 24 months of adjuvant hormonal therapy prior to recurrence.
The primary endpoint for the study was progression-free survival (PFS) evaluated by RECIST (Response Evaluation Criteria in Solid Tumors), based on the investigator's assessment (local radiology). Supportive PFS analyses were based on an independent central radiology review.
Secondary endpoints included overall survival (OS), objective response rate, clinical benefit rate, safety, change in quality of life (QoL) and time to ECOG PS (Eastern Cooperative Oncology Group performance status) deterioration.
A total of 724 patients were randomised in a 2:1 ratio to the combination everolimus (10 mg daily) + exemestane (25 mg daily) (n=485) or to the placebo + exemestane arm (25 mg daily) (n=239). At the time of the final OS analysis, the median duration of everolimus treatment was 24.0 weeks (range 1.0-199.1 weeks). The median duration of exemestane treatment was longer in the everolimus + exemestane group at 29.5 weeks (1.0-199.1) compared to 14.1 weeks (1.0-156.0) in the placebo + exemestane group.
The efficacy results for the primary endpoint were obtained from the final PFS analysis (see Table 4 and Figure 1). Patients in the placebo + exemestane arm did not cross over to everolimus at the time of progression.
Table 4. BOLERO-2 efficacy results:
| Analysis | Afinitora n=485 | Placeboa n=239 | Hazard ratio | p value |
|---|---|---|---|---|
| Median progression-free survival (months) (95% CI) | ||||
| Investigator radiological review | 7.8 (6.9 to 8.5) | 3.2 (2.8 to 4.1) | 0.45 (0.38 to 0.54) | <0.0001 |
| Independent radiological review | 11.0 (9.7 to 15.0) | 4.1 (2.9 to 5.6) | 0.38 (0.31 to 0.48) | <0.0001 |
| Median overall survival (months) (95% CI) | ||||
| Median overall survival | 31.0 (28.0 – 34.6) | 26.6 (22.6 – 33.1) | 0.89 (0.73 – 1.10) | 0.1426 |
| Best overall response (%) (95% CI) | ||||
| Objective response rateb | 12.6% (9.8 to 15.9) | 1.7% (0.5 to 4.2) | n/ad | <0.0001e |
| Clinical benefit ratec | 51.3% (46.8 to 55.9) | 26.4% (20.9 to 32.4) | n/ad | <0.0001e |
a Plus exemestane
b Objective response rate = proportion of patients with complete or partial response
c Clinical benefit rate = proportion of patients with complete or partial response or stable disease ≥24 weeks
d Not applicable
e p value is obtained from the exact Cochran-Mantel-Haenszel test using a stratified version of the Cochran-Armitage permutation test.
Figure 1. BOLERO-2 Kaplan-Meier progression-free survival curves (investigator radiological review):
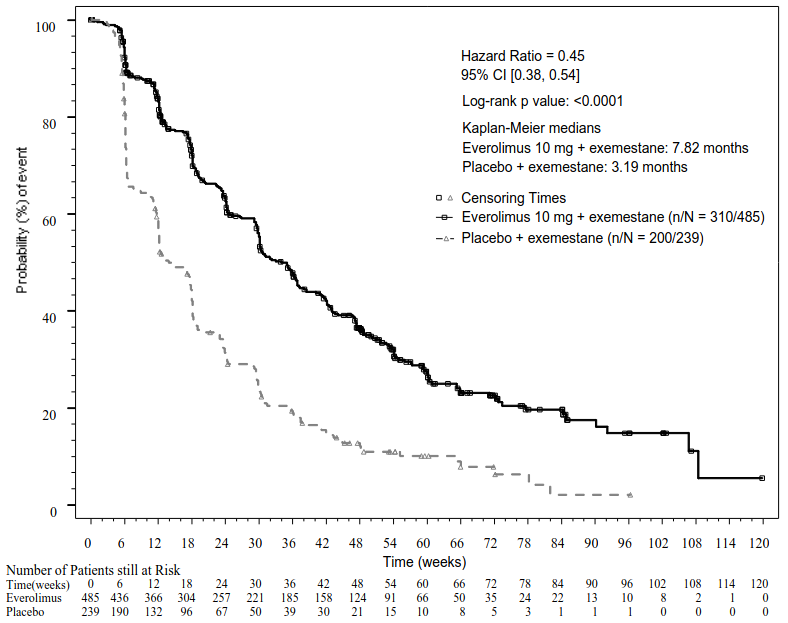
The estimated PFS treatment effect was supported by planned subgroup analysis of PFS per investigator assessment. For all analysed subgroups (age, sensitivity to prior hormonal therapy, number of organs involved, status of bone-only lesions at baseline and presence of visceral metastasis, and across major demographic and prognostic subgroups) a positive treatment effect was seen with everolimus + exemestane with an estimated hazard ratio (HR) versus placebo + exemestane ranging from 0.25 to 0.60.
No differences in the time to ≥5% deterioration in the global and functional domain scores of QLQ-C30 were observed in the two arms.
BOLERO-6 (Study CRAD001Y2201), a three-arm, randomised, open-label, phase II study of everolimus in combination with exemestane versus everolimus alone versus capecitabine in the treatment of postmenopausal women with oestrogen receptor-positive, HER2/neu negative, locally advanced, recurrent, or metastatic breast cancer after recurrence or progression on prior letrozole or anastrozole.
The primary objective of the study was to estimate the HR of PFS for everolimus + exemestane versus everolimus alone. The key secondary objective was to estimate the HR of PFS for everolimus + exemestane versus capecitabine.
Other secondary objectives included the evaluation of OS, objective response rate, clinical benefit rate, safety, time to ECOG performance deterioration, time to QoL deterioration, and treatment satisfaction (TSQM). No formal statistical comparisons were planned.
A total of 309 patients were randomised in a 1:1:1 ratio to the combination of everolimus (10 mg daily) + exemestane (25 mg daily) (n=104), everolimus alone (10 mg daily) (n=103), or capecitabine (1250 mg/m² dose twice daily for 2 weeks followed by one week rest, 3-week cycle) (n=102). At the time of data cut-off, the median duration of treatment was 27.5 weeks (range 2.0-165.7) in the everolimus + exemestane arm, 20 weeks (1.3-145.0) in the everolimus arm, and 26.7 weeks (1.4-177.1) in the capecitabine arm.
The result of the final PFS analysis with 154 PFS events observed based on local investigator assessment showed an estimated HR of 0.74 (90% CI: 0.57, 0.97) in favour of the everolimus + exemestane arm relative to everolimus arm. The median PFS was 8.4 months (90% CI: 6.6, 9.7) and 6.8 months (90% CI: 5.5, 7.2), respectively.
Figure 2. BOLERO-6 Kaplan-Meier progression-free survival curves (investigator radiological review):
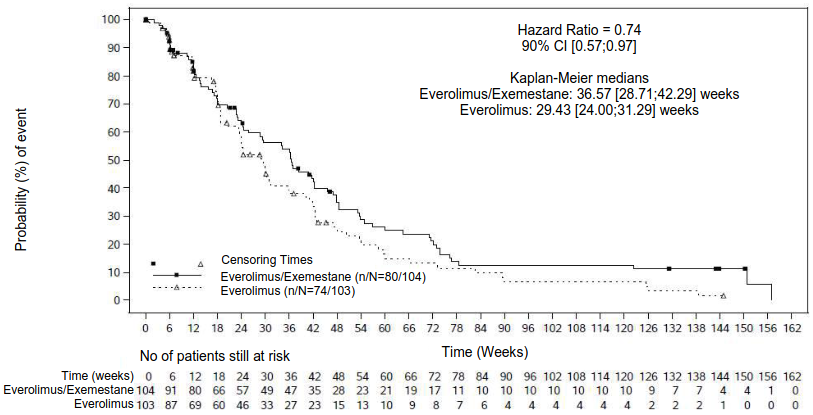
For the key secondary endpoint PFS the estimated HR was 1.26 (90% CI: 0.96, 1.66) in favour of capecitabine over the everolimus + exemestane combination arm based on a total of 148 PFS events observed.
Results of the secondary endpoint OS were not consistent with the primary endpoint PFS, with a trend observed favouring the everolimus alone arm. The estimated HR was 1.27 (90% CI: 0.95, 1.70) for the comparison of OS in the everolimus alone arm relative to the everolimus + exemestane arm. The estimated HR for the comparison of OS in the everolimus + exemestane combination arm relative to capecitabine arm was 1.33 (90% CI: 0.99, 1.79).
Advanced neuroendocrine tumours of pancreatic origin (pNET)
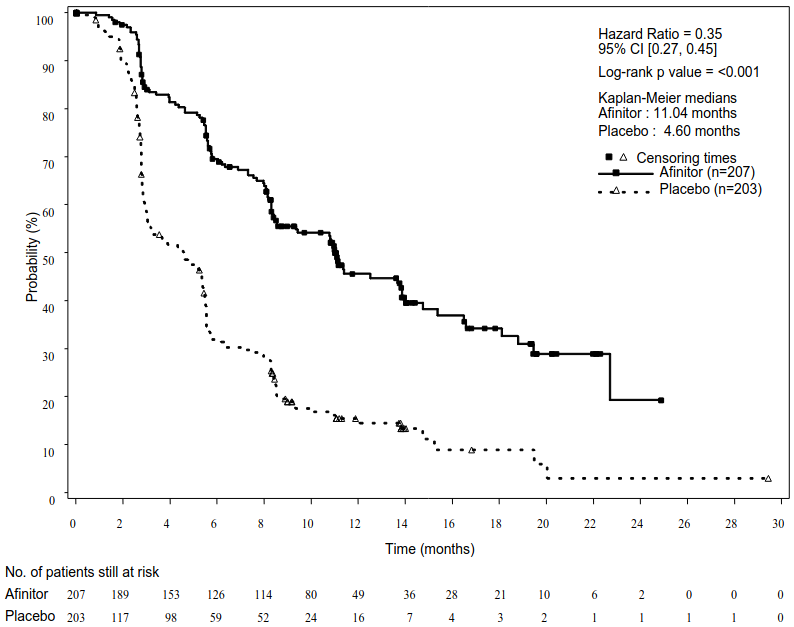
RADIANT-3 (study CRAD001C2324), a phase III, multicentre, randomised, double-blind study of Afinitor plus best supportive care (BSC) versus placebo plus BSC in patients with advanced pNET, demonstrated a statistically significant clinical benefit of Afinitor over placebo by a 2.4-fold prolongation of median progression-free-survival (PFS) (11.04 months versus 4.6 months), (HR 0.35; 95% CI: 0.27, 0.45; p<0.0001) (see Table 5 and Figure 3).
RADIANT-3 involved patients with well- and moderately-differentiated advanced pNET whose disease had progressed within the prior 12 months. Treatment with somatostatin analogues was allowed as part of BSC.
The primary endpoint for the study was PFS evaluated by RECIST (Response Evaluation Criteria in Solid Tumors). Following documented radiological progression, patients could be unblinded by the investigator. Those randomised to placebo were then able to receive open-label Afinitor.
Secondary endpoints included safety, objective response rate, response duration and overall survival (OS).
In total, 410 patients were randomised 1:1 to receive either Afinitor 10 mg/day (n=207) or placebo (n=203). Demographics were well balanced (median age 58 years, 55% male, 78.5% Caucasian). Fifty-eight percent of the patients in both arms received prior systemic therapy. The median duration of blinded study treatment was 37.8 weeks (range 1.1-129.9 weeks) for patients receiving everolimus and 16.1 weeks (range 0.4-147.0 weeks) for those receiving placebo.
Following disease progression or after study unblinding, 172 of the 203 patients (84.7%) initially randomised to placebo crossed over to open-label Afinitor. The median duration of open-label treatment was 47.7 weeks among all patients; 67.1 weeks in the 53 patients randomised to everolimus who switched to open-label everolimus and 44.1 weeks in the 172 patients randomised to placebo who switched to open-label everolimus.
Table 5. RADIANT-3 – efficacy results:
| Population | Afinitor n=207 | Placebo n=203 | Hazard ratio (95% CI) | p-value |
|---|---|---|---|---|
| Median progression-free survival (months) (95% CI) | ||||
| Investigator radiological review | 11.04 (8.41, 13.86) | 4.60 (3.06, 5.39) | 0.35 (0.27, 0.45) | <0.0001 |
| Independent radiological review | 13.67 (11.17, 18.79) | 5.68 (5.39, 8.31) | 0.38 (0.28, 0.51) | <0.0001 |
| Median overall survival (months) (95% CI) | ||||
| Median overall survival | 44.02 (35.61, 51.75) | 37.68 (29.14, 45.77) | 0.94 (0.73, 1.20) | 0.300 |
Figure 3. RADIANT-3 – Kaplan-Meier progression-free survival curves (investigator radiological review):
Advanced neuroendocrine tumours of gastrointestinal or lung origin
RADIANT-4 (study CRAD001T2302), a randomised, double-blind, multicentre, phase III study of Afinitor plus best supportive care (BSC) versus placebo plus BSC was conducted in patients with advanced, well-differentiated (Grade 1 or Grade 2) non-functional neuroendocrine tumours of gastrointestinal or lung origin without a history of and no active symptoms related to carcinoid syndrome.
The primary endpoint for the study was progression-free survival (PFS) evaluated by Response Evaluation Criteria in Solid Tumors (RECIST), based on independent radiology assessment. Supportive PFS analysis was based on local investigator review. Secondary endpoints included overall survival (OS), overall response rate, disease control rate, safety, change in quality of life (FACT-G) and time to World Health Organisation performance status (WHO PS) deterioration.
A total of 302 patients were randomised in a 2:1 ratio to receive either everolimus (10 mg daily) (n=205) or placebo (n=97). Demographics and disease characteristics were generally balanced (median age 63 years [range 22 to 86], 76% Caucasian, history of prior somatostatin analogue [SSA] use). The median duration of blinded treatment was 40.4 weeks for patients receiving Afinitor and 19.6 weeks for those receiving placebo. After primary PFS analysis, 6 patients from the placebo arm crossed over to open-label everolimus.
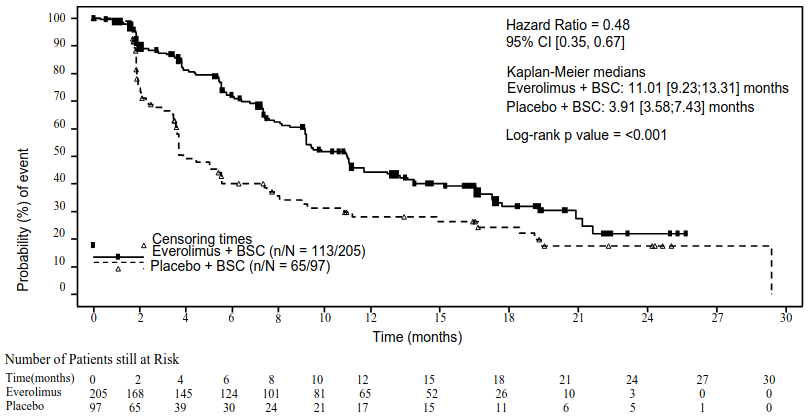
The efficacy results for the primary endpoint PFS (independent radiological review) were obtained from the final PFS analysis (see Table 6 and Figure 4). The efficacy results for PFS (investigator radiological review) were obtained from the final OS analysis (see Table 6).
Table 6. RADIANT-4 – Progression-free survival results:
| Population | Afinitor n=205 | Placebo n=97 | Hazard ratio (95% CI) | p-valuea |
|---|---|---|---|---|
| Median progression-free survival (months) (95% CI) | ||||
| Independent radiological review | 11.01 (9.2, 13.3) | 3.91 (3.6, 7.4) | 0.48 (0.35, 0.67) | <0.001 |
| Investigator radiological review | 14.39 (11.24, 17.97) | 5.45 (3.71, 7.39) | 0.40 (0.29, 0.55) | <0.001 |
a One-sided p-value from a stratified log-rank test
Figure 4. RADIANT-4 – Kaplan-Meier progression-free survival curves (independent radiological review):
In supportive analyses, positive treatment effect has been observed in all subgroups with the exception of the subgroup of patients with ileum as primary site of tumour origin (Ileum: HR=1.22 [95% CI: 0.56 to 2.65]; Non-ileum: HR=0.34 [95% CI: 0.22 to 0.54]; Lung: HR=0.43 [95% CI: 0.24 to 0.79]) (see Figure 5).
Figure 5. RADIANT-4 – Progression free survival results by pre-specified patient subgroup (independent radiological review):
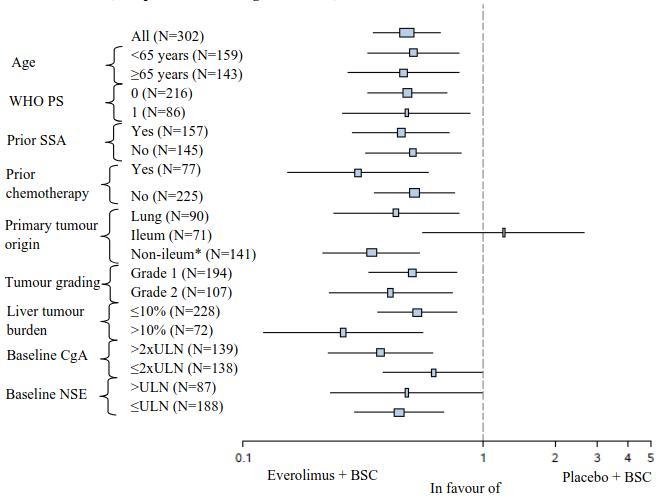
* Non-ileum: stomach, colon, rectum, appendix, caecum, duodenum, jejunum, carcinoma of unknown primary origin and other gastrointestinal origin
ULN: Upper limit of normal
CgA: Chromogranin A
NSE: Neuron specific enolase
Hazard ratio (95% CI) from stratified Cox model
The final overall survival (OS) analysis did not show a statistically significant difference between those patients who received Afinitor or placebo during the blinded treatment period of the study (HR=0.90 [95% CI: 0.66 to 1.22]).
No difference in the time to definitive deterioration of WHO PS (HR=1.02; [95% CI: 0.65, 1.61]) and time to definitive deterioration in quality of life (FACT-G total score HR=0.74; [95% CI: 0.50, 1.10]) was observed between the two arms.
Advanced renal cell carcinoma
RECORD-1 (study CRAD001C2240), a phase III, international, multicentre, randomised, double-blind study comparing everolimus 10 mg/day and placebo, both in conjunction with best supportive care, was conducted in patients with metastatic renal cell carcinoma whose disease had progressed on or after treatment with VEGFR-TKI (vascular endothelial growth factor receptor tyrosine kinase inhibitor) therapy (sunitinib, sorafenib, or both sunitinib and sorafenib). Prior therapy with bevacizumab and interferon-α was also permitted. Patients were stratified according to Memorial Sloan-Kettering Cancer Center (MSKCC) prognostic score (favourable- vs. intermediate- vs. poor-risk groups) and prior anticancer therapy (1 vs. 2 prior VEGFR-TKIs).
Progression-free survival, documented using RECIST (Response Evaluation Criteria in Solid Tumours) and assessed via a blinded, independent central review, was the primary endpoint. Secondary endpoints included safety, objective tumour response rate, overall survival, disease-related symptoms, and quality of life. After documented radiological progression, patients could be unblinded by the investigator: those randomised to placebo were then able to receive open-label everolimus 10 mg/day. The Independent Data Monitoring Committee recommended termination of this trial at the time of the second interim analysis as the primary endpoint had been met.
In total, 416 patients were randomised 2:1 to receive Afinitor (n=277) or placebo (n=139). Demographics were well balanced (pooled median age [61 years; range 27-85], 78% male, 88% Caucasian, number of prior VEGFR-TKI therapies [1-74%, 2-26%]). The median duration of blinded study treatment was 141 days (range 19-451 days) for patients receiving everolimus and 60 days (range 21-295 days) for those receiving placebo.
Afinitor was superior to placebo for the primary endpoint of progression-free survival, with a statistically significant 67% reduction in the risk of progression or death (see Table 7 and Figure 6).
Table 7. RECORD-1 – Progression-free survival results:
| Population | n | Afinitor n=277 | Placebo n=139 | Hazard ratio (95% CI) | p-value |
|---|---|---|---|---|---|
| Median progression-free survival (months) (95% CI) | |||||
| Primary analysis | |||||
| All (blinded independent central review) | 416 | 4.9 (4.0-5.5) | 1.9 (1.8-1.9) | 0.33 (0.25-0.43) | <0.0001a |
| Supportive/sensitivity analyses | |||||
| All (local review by investigator) | 416 | 5.5 (4.6-5.8) | 1.9 (1.8-2.2) | 0.32 (0.25-0.41) | <0.0001a |
| MSKCC prognostic score (blinded independent central review) | |||||
| Favourable risk | 120 | 5.8 (4.0-7.4) | 1.9 (1.9-2.8) | 0.31 (0.19-0.50) | <0.0001 |
| Intermediate risk | 235 | 4.5 (3.8-5.5) | 1.8 (1.8-1.9) | 0.32 (0.22-0.44) | <0.0001 |
| Poor risk | 61 | 3.6 (1.9-4.6) | 1.8 (1.8-3.6) | 0.44 (0.22-0.85) | 0.007 |
a Stratified log-rank test
Figure 6. RECORD-1 – Kaplan-Meier progression-free survival curves (independent central review):
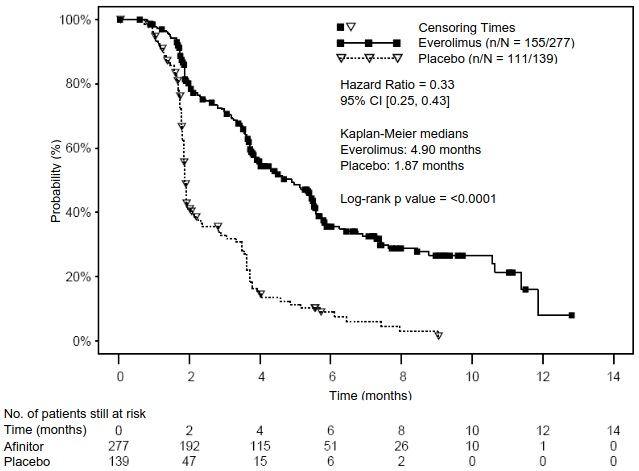
Six-month PFS rates were 36% for Afinitor therapy compared with 9% for placebo.
Confirmed objective tumour responses were observed in 5 patients (2%) receiving Afinitor, while none were observed in patients receiving placebo. Therefore, the progression-free survival advantage primarily reflects the population with disease stabilisation (corresponding to 67% of the Afinitor treatment group).
No statistically significant treatment-related difference in overall survival was noted (hazard ratio 0.87; confidence interval: 0.65-1.17; p=0.177). Crossover to open-label Afinitor following disease progression for patients allocated to placebo confounded the detection of any treatment-related difference in overall survival.
Other studies
Stomatitis is the most commonly reported adverse reaction in patients treated with Afinitor (see sections 4.4 and 4.8). In a post-marketing single-arm study in postmenopausal women with advanced breast cancer (N=92), topical treatment with dexamethasone 0.5 mg/5 ml alcohol-free oral solution was administered as a mouthwash (4 times daily for the initial 8 weeks of treatment) to patients at the time of initiating treatment with Afinitor (10 mg/day) plus exemestane (25 mg/day) to reduce the incidence and severity of stomatitis. The incidence of Grade ≥2 stomatitis at 8 weeks was 2.4% (n=2/85 evaluable patients) which was lower than historically reported. The incidence of Grade 1 stomatitis was 18.8% (n=16/85) and no cases of Grade 3 or 4 stomatitis were reported. The overall safety profile in this study was consistent with that established for everolimus in the oncology and tuberous sclerosis complex (TSC) settings, with the exception of a slightly increased frequency of oral candidiasis which was reported in 2.2% (n=2/92) of patients.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Afinitor in all subsets of the paediatric population in neuroendocrine tumours of pancreatic origin, thoracic neuroendocrine tumours and in renal cell carcinoma (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
In patients with advanced solid tumours, peak everolimus concentrations (Cmax) are reached at a median time of 1 hour after daily administration of 5 and 10 mg everolimus under fasting conditions or with a light fat-free snack. Cmax is dose-proportional between 5 and 10 mg. Everolimus is a substrate and moderate inhibitor of PgP.
Food effect
In healthy subjects, high fat meals reduced systemic exposure to everolimus 10 mg (as measured by AUC) by 22% and the peak plasma concentration Cmax by 54%. Light fat meals reduced AUC by 32% and Cmax by 42%. Food, however, had no apparent effect on the post absorption phase concentration-time profile.
Distribution
The blood-to-plasma ratio of everolimus, which is concentration-dependent over the range of 5 to 5,000 ng/ml, is 17% to 73%. Approximately 20% of the everolimus concentration in whole blood is confined to plasma in cancer patients given everolimus 10 mg/day. Plasma protein binding is approximately 74% both in healthy subjects and in patients with moderate hepatic impairment. In patients with advanced solid tumours, Vd was 191 l for the apparent central compartment and 517 l for the apparent peripheral compartment.
Biotransformation
Everolimus is a substrate of CYP3A4 and PgP. Following oral administration, everolimus is the main circulating component in human blood. Six main metabolites of everolimus have been detected in human blood, including three monohydroxylated metabolites, two hydrolytic ring-opened products, and a phosphatidylcholine conjugate of everolimus. These metabolites were also identified in animal species used in toxicity studies, and showed approximately 100 times less activity than everolimus itself. Hence, everolimus is considered to contribute the majority of the overall pharmacological activity.
Elimination
Mean oral clearance (CL/F) of everolimus after 10 mg daily dose in patients with advanced solid tumours was 24.5 l/h. The mean elimination half-life of everolimus is approximately 30 hours.
No specific excretion studies have been undertaken in cancer patients; however, data are available from the studies in transplant patients. Following the administration of a single dose of radiolabelled everolimus in conjunction with ciclosporin, 80% of the radioactivity was recovered from the faeces, while 5% was excreted in the urine. The parent substance was not detected in urine or faeces.
Steady-state pharmacokinetics
After administration of everolimus in patients with advanced solid tumours, steady-state AUC0-τ was dose-proportional over the range of 5 to 10 mg daily dose. Steady-state was achieved within 2 weeks. Cmax is dose-proportional between 5 and 10 mg. tmax occurs at 1 to 2 hours post-dose. There was a significant correlation between AUC0-τ and pre-dose trough concentration at steady-state.
Special populations
Hepatic impairment
The safety, tolerability and pharmacokinetics of everolimus were evaluated in two single oral dose studies of Afinitor tablets in 8 and 34 subjects with impaired hepatic function relative to subjects with normal hepatic function.
In the first study, the average AUC of everolimus in 8 subjects with moderate hepatic impairment (Child-Pugh B) was twice that found in 8 subjects with normal hepatic function.
In the second study of 34 subjects with different impaired hepatic function compared to normal subjects, there was a 1.6-fold, 3.3-fold and 3.6-fold increase in exposure (i.e. AUC0-inf) for subjects with mild (Child-Pugh A), moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment, respectively.
Simulations of multiple dose pharmacokinetics support the dosing recommendations in subjects with hepatic impairment based on their Child-Pugh status.
Based on the results of the two studies, dose adjustment is recommended for patients with hepatic impairment (see sections 4.2 and 4.4).
Renal impairment
In a population pharmacokinetic analysis of 170 patients with advanced solid tumours, no significant influence of creatinine clearance (25-178 ml/min) was detected on CL/F of everolimus. Post-transplant renal impairment (creatinine clearance range 11-107 ml/min) did not affect the pharmacokinetics of everolimus in transplant patients.
Elderly patients
In a population pharmacokinetic evaluation in cancer patients, no significant influence of age (27-85 years) on oral clearance of everolimus was detected.
Ethnicity
Oral clearance (CL/F) is similar in Japanese and Caucasian cancer patients with similar liver functions. Based on analysis of population pharmacokinetics, CL/F is on average 20% higher in black transplant patients.
5.3. Preclinical safety data
The preclinical safety profile of everolimus was assessed in mice, rats, minipigs, monkeys and rabbits. The major target organs were male and female reproductive systems (testicular tubular degeneration, reduced sperm content in epididymides and uterine atrophy) in several species; lungs (increased alveolar macrophages) in rats and mice; pancreas (degranulation and vacuolation of exocrine cells in monkeys and minipigs, respectively, and degeneration of islet cells in monkeys), and eyes (lenticular anterior suture line opacities) in rats only. Minor kidney changes were seen in the rat (exacerbation of age-related lipofuscin in tubular epithelium, increases in hydronephrosis) and mouse (exacerbation of background lesions). There was no indication of kidney toxicity in monkeys or minipigs.
Everolimus appeared to spontaneously exacerbate background diseases (chronic myocarditis in rats, coxsackie virus infection of plasma and heart in monkeys, coccidian infestation of the gastrointestinal tract in minipigs, skin lesions in mice and monkeys). These findings were generally observed at systemic exposure levels within the range of therapeutic exposure or above, with the exception of the findings in rats, which occurred below therapeutic exposure due to a high tissue distribution.
In a male fertility study in rats, testicular morphology was affected at 0.5 mg/kg and above, and sperm motility, sperm head count, and plasma testosterone levels were diminished at 5 mg/kg which caused a reduction in male fertility. There was evidence of reversibility.
In animal reproductive studies female fertility was not affected. However, oral doses of everolimus in female rats at ≥0.1 mg/kg (approximately 4% of the AUC0-24h in patients receiving the 10 mg daily dose) resulted in increases in pre-implantation loss.
Everolimus crossed the placenta and was toxic to the foetus. In rats, everolimus caused embryo/foetotoxicity at systemic exposure below the therapeutic level. This was manifested as mortality and reduced foetal weight. The incidence of skeletal variations and malformations (e.g. sternal cleft) was increased at 0.3 and 0.9 mg/kg. In rabbits, embryotoxicity was evident in an increase in late resorptions.
Genotoxicity studies covering relevant genotoxicity endpoints showed no evidence of clastogenic or mutagenic activity. Administration of everolimus for up to 2 years did not indicate any oncogenic potential in mice and rats up to the highest doses, corresponding respectively to 3.9 and 0.2 times the estimated clinical exposure.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.