ANDROCUR Tablet / Solution for injection Ref.[50194] Active ingredients: Cyproterone
Source: Health Products and Food Branch (CA) Revision Year: 2021
Action and clinical pharmacology
10.1 Mechanism of Action
ANDROCUR (cyproterone acetate) is a steroid which clinically demonstrates two distinct properties:
a) Antiandrogenic: Cyproterone acetate blocks the binding of dihydrotestosterone – the active metabolite of testosterone – to the specific receptors in the prostatic carcinoma cell.
b) Progestogenic/antigonadotrophic: Cyproterone acetate exerts a negative feed-back on the hypothalamo-pituitary axis, by inhibiting the secretion of LH leading to diminished production of testicular testosterone.
10.3 Pharmacokinetics
Absorption
ANDROCUR
The absorption of cyproterone acetate following oral administration is complete. Peak plasma levels are reached 3 to 4 hours after administration. Plasma levels fall rapidly during the first 24 hours as a result of tissue distribution and excretion, and plasma half-life was 38 ± 5 hours.
ANDROCUR DEPOT
Following intramuscular administration, mean maximum blood levels are attained 3.4 days after injection. The mean elimination half-life was found to be 4 days.
Metabolism
The principal metabolite identified was 15β-hydroxy-cyproterone acetate.
Excretion
Most of the cyproterone acetate is excreted unchanged in the feces (60%) or urine (33%) within 72 hours.
Cyproterone acetate is eliminated with the urine mainly in the form of unconjugated metabolites and with the bile (feces) in the form of glucuronidized metabolites.
Detailed Human Pharmacology
Antiandrogenic Effect
The following actions which are associated with the antiandrogenic effects have been described in man: reduction of sexual drive; inhibition of spermatogenesis; palliative effect in prostatic carcinoma; inhibition of sebaceous gland activity; suppression of signs of androgenization in women; inhibition of premature genital development in children; and other associated symptoms.
Progestogenic and Antigonadotrophic Effect
Cyproterone acetate in man is also a potent progestogen and has an antigonadotrophic effect. It intervenes with the hypothalamo-pituitary pathway, causing an inhibition of increased secretion of LH, and a decrease in gonadal testicular androgens.
Thus, unlike pure antiandrogens, cyproterone acetate does not cause a compensatory increase in androgen secretion.
Other Endocrine Effects
No distinct influence on the 17-ketosteroids, 17-ketogenic steroids or on total estrogens in the 24-hour urine has been observed in male patients. On fluorometric determination of urinary cortisol, the value apparently increases because the cyproterone acetate eliminated with the urine is also measured. Simultaneously, cyproterone acetate also reduces the reaction of the adrenal cortex to exogenous ACTH in patients; the baseline cortisol and ACTH values may also be reduced.
Pharmacokinetics
A bioavailability study was performed in 5 male volunteer subjects receiving a single oral dose of 50 mg 14C-cyproterone acetate tablets.
Results of the study showed that cyproterone acetate is absorbed slowly, but completely (100%), from the gastrointestinal tract. The maximum plasma level was reached 3 to 4 hours after ingestion. The mean plasma levels were 700 nmol/L (=290µg/L) cyproterone acetate or, including the radioactivity of metabolites, 960 nmol/L (=400µg/L) cyproterone acetate equivalent.
The plasma levels fell quickly up to 24 hours after administration because of extensive tissue distribution. The half-life of cyproterone acetate in plasma was calculated as 38 ± 5 hours (see Figure 1).
Figure 1. Relationship of Unchanged Cyproterone Acetate to the Total 14C-labelled Substance (Cyproterone Acetate + Metabolites) in the Plasma of a Male Subject Following Oral Administration of 50 mg 14C-cyproterone Acetate:

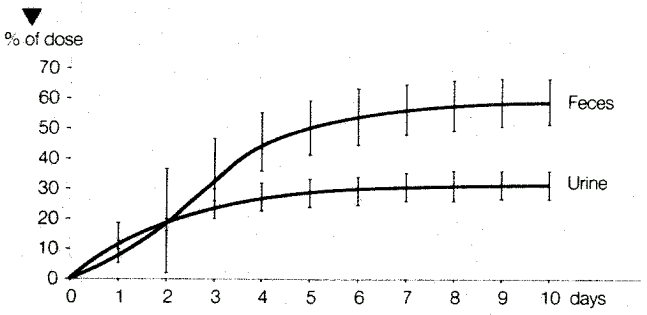
On oral administration cyproterone acetate was eliminated with a half-life of 38 ± 2 hours. After 10 days, 33 ± 6% of the dose could be recovered in the urine and 60 ± 8% in the feces (see Figure 2).
Figure 2. Elimination (% of Dose) Following Oral Administration of 50 mg 14C-cyproterone Acetate in Male Subjects. Mean Values ± Standard Deviation (n=5):
The intramuscular injection of 300 mg radioactivity labelled cyproterone acetate in a castor oil solution corresponding to ANDROCUR DEPOT was administered to male patients.
The maximum plasma level was reached 82 ± 21 hours after administration. Half the maximum value could be seen less than 24 hours after administration; the values did not fall below this level until about 10 days after dosing (see Figure 3).
Figure 3. Plasma Levels (Cyproterone Acetate Equivalents/mL) Following IM injection of 300 mg Cyproterone Acetate in Oily Solution in Male Patients. Mean Values ± Standard Deviation (n=11):

The elimination half-life of the cyproterone acetate released from the depot was 38 ± 14 hours which is the same as measured under oral administration.
A steady-state study was also carried out in 5 patients who received 300 mg of ANDROCUR DEPOT on a weekly basis. Determinations of the cyproterone acetate concentrations were carried out after the first, third, and fifth injections. Table 2 summarizes the results of the study.
In man, cyproterone acetate is eliminated in the urine mainly in the form of unconjugated metabolites and in the bile in the form of glucuronized metabolites; the main metabolite was 15β-OH cyproterone acetate.
Table 2. Pharmacokinetic Parameters After One and Several Intramuscular Injections of 300 mg Cyproterone Acetate in an Oil Solution (ANDROCUR DEPOT) in 5 Patients (Mean Values ± SD):
| Parameter | 1st Injection | 3rd Injection | 5th Injection |
|---|---|---|---|
| tmax (d) | 1.8 ± 0.4 | 2.4 ± 0.5 | 3.0 ± 1.0 |
| Cmax (ng/mL) | 273 ± 54 | 387 ± 111 | 406 ± 57 |
| t½ (d) | 4.4 ± 1.9 | 4.1 ± 1.3 | 3.9 ± 1.3 |
Detailed pharmacology
A total of 24 studies have been conducted with ANDROCUR (cyproterone acetate) in patients requiring palliative treatment for advanced prostatic carcinoma. Worldwide, more than 1,000 patients have participated in these studies, which included several large multicentre trials in addition to the important comparative multicentre trial conducted by the European Cancer Oncology Group. North American experience has been accumulated in the U.S. by Drs. Scott (Johns Hopkins Hospital, Baltimore), Geller (Mercy Hospital & Medical Center, San Diego), and by Drs. Wein and Murphy (Hospital of the University of Pennsylvania, Philadelphia).
14.1 Trial Design and Study Demographics
Patients and Stage of Disease
As shown in Table 3, more than 90% of the patients treated with ANDROCUR had stage C advanced prostatic carcinoma, or stage D1 or D2 prostatic carcinoma with metastasis.
Table 3. Patients:
| Stage | No. of Patients |
|---|---|
| A or B | 18 |
| C | 174 |
| C or D | 502 |
| D | 349 |
| Not specified | 39 |
| Total | 1082 |
The majority of patients (75%) had had no therapy prior to treatment with ANDROCUR. A large group of patients had received various types of estrogen therapy, but had proven to be refractory or unable to tolerate the drug. A few patients had undergone an orchiectomy or had received radiation therapy (Table 4).
Table 4. Previous Therapy:
| Previous Therapy | No. of Patients |
|---|---|
| None | 809 |
| Orchiectomy | 76 |
| Estrogen | 253 |
| Radiation | 16 |
Dosage and Administration
The oral route of administration of ANDROCUR was employed in 910 patients (84%), while 172 patients received ANDROCUR DEPOT, an oily solution containing 100 mg/mL cyproterone acetate. The standard dose of the latter was one weekly IM injection of 300 mg. As shown in the table below (Table 5), the daily oral dose varied considerably from study to study and from patient to patient. However, most patients were treated with doses ranging from 200 to 300 mg/day. In orchiectomized patients, the daily dose was generally reduced by about 50% to a range of 100 to 200 mg/day orally or the frequency of ANDROCUR DEPOT injections was reduced to one every 2 weeks.
Table 5. Dose of ANDROCUR or ANDROCUR DEPOT:
| Entity | Route | Dose | No. of Patients |
|---|---|---|---|
| ANDROCUR | Oral | 100 mg/day | 15 |
| 200 mg/day | 197 | ||
| 250 mg/day | 135 | ||
| 300 mg/day | 114 | ||
| 100-300 mg/day | 449 | ||
| ANDROCUR DEPOT | IM | 300 mg/week | 172 |
Only 32 patients (3%) received concomitant drug therapy with ANDROCUR. No other patients received concomitant drugs, but 521 patients (48%) underwent an orchiectomy (Table 6).
Table 6. Concomitant Therapy:
| Concomitant Therapy | No. of Patients |
|---|---|
| None | 529 |
| Estrogen (DES 0.1 mg) | 32 |
| Orchiectomy | 521 |
14.2 Study Results
Effect on Serum Testosterone and Prostatic Acid Phosphatase (PAP)
Table 7. Effect on Serum Testosterone and Prostatic Acid Phosphatase (PAP):
| Parameter | No. of Studies | Result |
|---|---|---|
| Serum testosterone | 7 | 70-90% reduction |
| Prostatic acid phosphatase | 11 | Normalization in 90% of responding patients |
The effect of ANDROCUR on serum testosterone was monitored in 7 studies (Table 7). Serum testosterone was rapidly reduced following daily oral doses of 200 to 300 mg, with castrate levels being achieved within 1 to 4 weeks. The reduction was usually in the order of 70% to 90%; the greatest percent reduction occurred when ANDROCUR was combined with estrogen.
Results of PAP evaluations consistently showed a normalization of values within a very short time in responding patients. Similarly, when there were signs of progressing metastasis, PAP values again deviated from normal levels.
Effect on Primary Tumor
The effect of ANDROCUR on the primary tumor was assessed in a total of 678 patients. Of these, 489 were previously untreated; the primary tumor was reduced in 318 of these (65%) and was stabilized in another 69 (14%). Thus, the overall positive response rate in this group was 79% (Table 8).
A significant, though smaller, percentage (59%) of estrogen-refractory patients also exhibited a positive result.
Table 8. Effect on Primary Tumor:
| Patient Group | Number | Response of Primary Tumor | Total With Positive Effect | |
|---|---|---|---|---|
| Reduced | Stabilized | |||
| Previously untreated | 489 | 318 (65%) | 69 (14%) | 387 (79%) |
| Estrogen refractory | 189 | 112 (59%) | - | 112 (59%) |
Effect on Metastasis
As shown in Table 9, metastasis was reduced in 31% of 216 evaluable patients who had not previously been treated, but in only 13% of the evaluable estrogen-refractory patients. The progression of metastases appeared to be time-dependent. Despite reduced serum testosterone levels, metastases progressed over a period of several months to years, even in patients who were initially stabilized. The major cause of death during therapy with ANDROCUR was the progression of metastases and not the primary tumors.
Table 9. Effect on Metastases:
| Patient Group | Number | Response of Metastases | Total with Positive Effect | |
|---|---|---|---|---|
| Reduced | Stabilized | |||
| Previously untreated | 216 | 67 (31%) | 82 (39%) | 149 (70%) |
| Estrogen refractory | 71 | 10 (13%) | 7 (10%) | 17 (23%) |
Effect on Pain
Table 10 illustrates the incidence of pain relief reported in each of 13 studies. Pain relief was noted in approximately 50% to 80% of patients receiving treatment with ANDROCUR. The effect of ANDROCUR on pain generally paralleled its effect on metastases. As long as metastases remained improved or stabilized, the analgesic requirement was also reduced. Renewed analgesic requirements were frequently indicative of metastatic progression.
Table 10. Pain Relief:
| Investigator | Incidence of Pain Relief |
|---|---|
| Dr. Bracci | 172/216 |
| Dr. Giuliani | 12/16 |
| Dr. Smith | 12/25 |
| Dr. Scott | 8/10 |
| Dr. Geller | 8/10 |
| Dr. Mauermayer | 38/58 |
| Dr. Wein | 13/24 |
| Dr. Tveter | 2/6 |
| Dr. Di Silverio | 13/20 |
| Dr. Ah-Lan | 9/16 |
| Dr. Pescatore | 12/16 |
| Dr. Hermabessiere | 2/4 |
| Dr. Bruchovsky | 15/24 |
| Total | 316/425 = 74% |
Subjective and Objective Responses
A general improvement in the subjective assessment of the quality of life was achieved in 70% of the 367 evaluable patients (Table 11).
The objective evaluations of remissions shown in Table 11 were based on ECOG criteria. The best results were obtained when ANDROCUR was used in combination with orchiectomy. One study revealed that more than ⅓ of the patients treated with ANDROCUR achieved a complete or partial remission for 3 to 5 years. The Canadian study found that a complete or partial remission was still evident in 75% of the patients after one year of treatment.
Table 11. Subjective and Objective Responses:
| Subjective Responses | |||
|---|---|---|---|
| No. Evaluable Patients | No. Improveda | ||
| 367 | 255 (70%) | ||
| Objective Responses (ECOG Criteria) | |||
| Treatment | Patient Group | No. of Patients | No. With Complete or Partial Remissions |
| ANDROCUR | Previously untreated | 270 | 134 (50%) |
| ANDROCUR | Estrogen-refractory | 77 | 31 (44%) |
| ANDROCUR/ Orchiectomy | Previously untreated and/or estrogen- refractory | 274 | 154 (60%) |
a Based on criteria of general improvement in quality of life (ie, weight gain, pain relief, etc.)
Survival Rate
Table 12. Survival Rate:
| Investigator | No. of Patients | Stage | Duration of Treatment | Survival | |
|---|---|---|---|---|---|
| ANDROCUR | Estrogen | ||||
| Dr. Mauermayer | 58 | C or D | 2-5 years | 38/58 (70%) | - |
| Dr. Wein | 55 | A (7) | 4 years | 39/55 (70%) | - |
| C (25) | |||||
| D (23) | |||||
| Dr. Bracci | 216 | C or D | 5 years | 138/216 (64%) | - |
| Dr. Di Silverio | 20 | D | up to 38 months | 3/20 (15%) | - |
| Dr. Giuliani | 68 | C | 5 years | 30/68 (44%) | 31% |
| Dr. Giuliani | 38 | D | 3 years | 10/38 (27%) | 10% |
| Dr. Jacobi | 51 | C or D | 2 years | 18/40 (45%) | - |
| Dr. Pavone | 103 | C or D | 3.5-5 years | 42/103 (41%) | 41% |
| Dr. Bruchovsky | 29 | D | 9-15 months | 23/29 (80%) | - |
As shown in Table 12 above, 5-year survival rates ranged from 41% to 64%. The 3-year rate for stage D patients was 27% and 1- to 2-year rates varied from a low of 15% up to a high of 80%. These survival rates generally represented an improvement over results previously obtained with estrogen therapy.
Microbiology
No microbiological information is required for this drug product.
Toxicology
General Toxicology
ANDROCUR (cyproterone acetate) has been found at low doses of 2 to 10 mg/kg to cause liver abnormalities in dogs and rats in the form of proliferative liver changes including increased liver weight, liver cell hypertrophy with an increase in the smooth endoplasmic reticulum, and a rise in the serum glutamic pyruvic transaminase (SGPT). At high doses of 50 to 100 mg/kg, nodular hepatic hyperplasia and hepatomas have also been observed.
In repeat-dose studies conducted in rats (12 weeks) and dogs (54 weeks) with oral administration of cyproterone acetate, decreased adrenal weights in rats at 4.0 mg/kg/day and dogs at 10 mg/kg were noted. Marked atrophy of zona fasciculate and of zona reticularis with preservation of zona glomerulosa was also observed in the adrenal glands of all treated dogs.
Acute Toxicity
The LD50 after single application of cyproterone acetate was as follows:
Table 13. LD50 After Single Application of Cyroterone Acetate:
| Animal Species | Oral (mg/kg) | Subcutaneous (mg/kg) | Intraperitoneal (mg/kg) | Intramuscular (mg/kg) |
|---|---|---|---|---|
| Mouse | >6000 | >5000 | >4000 | - |
| Rat | >4000 | 1500 | 1000 | - |
| Dog | >3000 | - | - | >100 (approx.) |
On the basis of the above LD50 values, cyproterone acetate can be considered practically nontoxic following single dose administration. The maximum intramuscular doses were also tolerated without symptoms in the dog, with exception of local tolerance manifestation.
Repeated Dose Toxicity
Repeat-dose toxicity studies revealed pathological changes in the liver, reproductive organs, adrenal glands, abnormal laboratory tests, and neoplasms of various tissues and organs in the animal species tested.
Chronic Toxicity Studies
Table 14. Chronic Toxicity Studies:
| Animal Species | Dosage and Duration | Mortality and Clinical and Laboratory Observations | Necropsy and Histopathology |
|---|---|---|---|
| Rats 35/sex/dose | 0; 10; 50, and 250 mg/kg 78 weeks oral | 250 mg/kg: marked increase in mortality rate. 50 and 250 mg/kg: 40-50% decrease in body weight gain. SGPT increase: males 10 and 250 mg/kg; females 50 mg/kg. BUN increase: males 50 and 250 mg/kg. Cholesterol increase: all treated groups. | Dose-related increase in liver weights. Increase thyroid weight except for low dose males. Dose-related decrease in gonads, adrenal, prostate, seminal vesicle, and uterus weights. Histopathology: toxic manifestation in liver and kidneys - less at 10 mg/kg, more extensive at 50 and 250 mg/kg. Changesincluded: yellow nodules and mottling of liver (including liver cell hyperplasia and liver cell adenomas and endoplasmic inclusion bodies), discolored kidneys with rough surfaces. |
| Rats 60/sex/dose | 0; 0.04; 0.4, and 2 mg/kg 104 weeks oral | No drug-relatedmortality. Dose-related decrease in body weight gains in males and increase in females. Food consumption reduced and thinning and loss of hair was also noted for high-dose males. Decrease in hemoglobin and erythrocytes at 0.4 and 2 mg/kg. SGOT, SGPT and alkaline phosphatase increased at 2 mg/kg. | 2 mg/kg increased incidence of subcutaneous masses and/or nodules; liver discoloration and nodules; atrophy of testes, seminal vesicles, and prostate. Increased incidence of mammary neoplasms (adenomas and adenocarcinomas). |
| Mice 50/sex/dose | 0; 0.04; 0.4, and 2 mg/kg 105 weeks oral | No dose-related mortality. Thinning and loss of hair at 2 mg/kg. Slightly reduced body weight gain at 2 mg/kg. | Slightly increased incidence of skin masses and/or nodules and alopecia. No drug-related inflammatory, degenerative, proliferative and/or neoplastic lesions. |
| Dogs Beagle 4/sex/dose | 0; 10; 32, and 100 mg/kg 55 weeks oral | No mortality. Excessive lacrimation, retarded pupillary reflex, mild conjunctivitis, hyperemia of gums, abdominal distention, sparsity of hair, and quieted behaviour. Laboratory tests: slightly elevated alkaline phosphatase and SGPT at 100 mg/kg in 2 dogs. Elevated sedimention rate, slightly reduced lymphocytes with increase in segmented neutrophils and decrease in eosinophils. | Reduced adrenal, testes, and prostate weight for all cyproterone acetate-treated animals. Ovary and uterus weights reduced at 100 mg/kg. Liver weight slightly increased for some dogs. Histopathology: marked adrenal atrophy of zona fasciculata and reticularis, testicular atrophy and absence of spermatogenesis, some Leydig cell hyperplasia, prostatic atrophy, ovarian and uterine atrophy, hyperplasia of mammary gland in males and females. |
| Rhesus monkey 4 females/ dose | 0; 0.04; 0.4, and 40 mg/kg 12 weeks oral | No mortality or behaviour changes. Dose-related alopecia. Raised insulin level above 0.04 mg/kg. Negative influence on coagulation at 0.4 mg/kg and 40 mg/kg. Stimulation of ACTH cells at 0.4 mg and above. Increase in prolactin cells and slight reduction in gonadotrophin cells. Galactorrhea in all treated. | At doses of 0.4 mg/kg and above - diffuse liver cell hypertrophy and an increase in smooth endoplasmic reticulum. At the two highest doses, 2 and 3 animals also had occasional eosinophil cytoplasmic inclusion bodies in the liver cells. In most treated animals small mammary nodules were palpable in the glandular tissue; at 40 mg/kg slight ductus proliferation was also noted. |
Mutagenesis and Carcinogenesis
Recognized first-line tests of genotoxicity gave negative results when conducted with CPA. No mutagenic effect of cyproterone acetate was demonstrated in either in vitro (Salmonella typhimurium) or in vivo (micronucleus test in the monkey). However, further tests showed that CPA was capable of producing DNA adducts and an increase in DNA repair activity in liver cells from rats and monkeys and also in freshly isolated human hepatocytes.
This DNA-adduct formation occurred at systemic exposures that might be expected to occur in the recommended dose regimens for CPA. One in vivo consequence of CPA treatment was the increased incidence of focal, possible preneoplastic liver lesions in which cellular enzymes were altered in female rats. An initiating potential besides promoting effect of cyproterone acetate on the formation of ATPase deficient and γGT positive foci in female rat livers was noted. CPA also enhanced the frequencies of mutations in the livers of female transgenic rats in a dose-dependent manner, indicating that CPA is mutagenic.
Investigations into the tumorigenicity of cyproterone acetate did not reveal a specific tumorigenic potential in the liver of rodents although other neoplasms, including mammary adenocarcinoma in rats, were observed (see Table 14).
Reproductive Toxicology
Testicular atrophy and absence of spermatogenesis, some Leydig cell hyperplasia, prostatic atrophy, ovarian and uterine atrophy were observed in beagle dogs. A reduced number of pregnancies in untreated female rats was observed when male rats were administered with 40 mg/kg/day cyproterone acetate. The temporary inhibition of fertility in male rats brought about by daily oral treatment of cyproterone acetate did not result in malformations or impairment of fertility in the offspring produced by untreated female animals.
The treatment of pregnant animals with cyproterone acetate leads to developmental disturbances in male fetuses. Testosterone-dependent differentiation processes are affected: signs of feminization of varying degrees of severity develop.
Table 15. Fertility and Reproduction Study:
| Animal Species | Route and Dosage of Administration | Findings |
|---|---|---|
| Rats 24/sex/dose (2 generations) | 0; 0.4; 4.0 and 40 mg/kg oral | 0.4 mg/kg: No influence by drug on fertility of the P1 and F1 generations. 4 mg/kg: Significant decrease in body weights but no impairment of preand postnatal development. 40 mg/kg: Food intake and body weight gain reduced. Although attempted matings were increased, less than 50% of the females had litters. No specific pathological changes were found in the dams, fetuses, or young. Similarly, no malformations were observed. |
Detailed Animal Pharmacology
Antiandrogenic Effects
Cyproterone acetate at doses of 10 or 50 mg/kg inhibits the effects of endogenously produced and exogenously administered androgens at the prostate by means of competitive inhibition.
In mice and dogs, cyproterone acetate induces a dose-dependent atrophy of the accessory sex glands, the prostate, seminal vesicles, and preputial glands. Spermatogenesis is inhibited in a dose-related manner; however, the atrophy in the Leydig cells are slight.
In the rat the start of puberty is prevented or delayed. Cyproterone acetate inhibits the physiological closure of the epiphyseal cartilages and bone maturation.
It impairs the function of the sebaceous glands, and the thickness of the epidermis decreases.
The treatment of pregnant animals with cyproterone acetate leads to developmental disturbances in male fetuses. Testosterone-dependent differentiation processes are affected: signs of feminization of varying degrees of severity develop.
Progestogenic and Antigonadotrophic Effect
On subcutaneous injections a total dose of 0.003 mg cyproterone acetate is about 100 times stronger than progesterone in the maintenance of pregnancy (Clauberg test). Like all potent progestogens, cyproterone acetate has antigonadotrophic properties which can be demonstrated in the parabiosis test, the testicular inhibition test in infantile rats, and by the inhibition of ovulation.
Pharmacokinetic Studies in Animals
Pharmacokinetic studies have been carried out in a number of animal species (rats, rabbits, dogs, and monkeys) using either methylene14C-or carboxy14C-labelled cyproterone acetate.
Cyproterone acetate is absorbed at most dose levels tested except in high doses. Peak plasma levels are usually obtained within 1 to 4 hours of oral dosing. Because of its lipophilic character, cyproterone acetate is taken up and concentrated in the liver and fatty tissues in all animal species. Cyproterone acetate is not hydrolysed, and mainly cyproterone acetate and the metabolite 15β-hydroxycyproterone acetate are found in the tissues and in plasma. The elimination half-life of cyproterone acetate is slow in most species (1-2 days), in a ratio of 4:6 with urine and feces; an exception is the dog, which excretes cyproterone acetate in 1 to 3 days. On repeated daily dosing, cyproterone acetate shows limited rise, and plasma levels can be taken as a reliable index of the concentrations of cyproterone acetate in the body. Cyproterone acetate passes the placental barrier, but only reaches the fetus in low concentrations. The pharmacokinetics, biotransformation, and metabolic spectra of cyproterone acetate are similar in man and the rhesus monkey.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.