AQUIPTA Tablet Ref.[51145] Active ingredients: Atogepant
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061 Ludwigshafen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Analgesics, calcitonin gene-related peptide (CGRP) antagonists
ATC code: N02CD07
Mechanism of action
Non-clinical receptor binding studies and in vitro functional studies point to an involvement of more than one receptor type in the pharmacological effects of atogepant. Atogepant shows affinity to several receptors of the calcitonin/CGRP-receptor family. In view of the clinically relevant free plasma concentrations of atogepant (Cmax >20 nM for a 60 mg dose) and the fact that CGRP and amylin-1 receptors are considered to be involved in the pathophysiology of migraine, inhibitory effects of atogepant at these receptors (Ki-value 26 pM and 2.4 nM, respectively) could be of clinical relevance. However, the precise mechanism of action of atogepant in the prophylaxis of migraine remains to be established.
Clinical efficacy and safety
Atogepant was evaluated for the prophylaxis of migraine in two pivotal studies across the migraine spectrum in chronic and episodic migraine. The episodic migraine study (ADVANCE) enrolled patients who met International Classification of Headache Disorders (ICHD) criteria for a diagnosis of migraine with or without aura. The chronic migraine study (PROGRESS) enrolled patients who also met ICHD criteria for chronic migraine. Both studies excluded patients with myocardial infarction, stroke, or transient ischemic attacks within six months prior to screening.
Episodic migraine
Atogepant was evaluated for the prophylaxis of episodic migraine (4 to 14 migraine days per month) in a randomised, multicentre, double-blind, placebo-controlled study (ADVANCE). Patients were randomised to AQUIPTA 60 mg (N=235) or placebo (N=223) once daily for 12 weeks. Patients were allowed to use acute headache treatments (i.e., triptans, ergotamine derivatives, NSAIDs, paracetamol and opioids) as needed. The use of a concomitant medicinal product that acts on the CGRP pathway was not permitted for either acute or preventive treatment of migraine.
A total of 88% patients completed the 12-week double-blind study period. Patients had a mean age of 42 years (range: 18 to 73 years), 4% were 65 years or older, 89% were female, and 83% were white. The mean migraine frequency at baseline was approximately 8 migraine days per month and was similar across treatment groups.
The primary efficacy endpoint was the change from baseline in mean monthly migraine days (MMD) across the 12-week treatment period. Secondary endpoints controlled for multiplicity included the change from baseline in mean monthly headache days, the change from baseline in mean monthly acute medication use days, the proportion of patients achieving at least a 50% reduction from baseline in mean MMD (3 month average), and several patient-reported outcome measures assessing functioning. Statistically significant findings were demonstrated for AQUIPTA versus placebo for the primary and secondary efficacy endpoints in ADVANCE, as summarized in Table 3.
Table 3. Efficacy endpoints in ADVANCE:
| AQUIPTA 60 mg N=226 | Placebo N=216 | |
|---|---|---|
| Monthly migraine days (MMD) across 12 weeks | ||
| Baseline | 7.8 | 7.5 |
| Mean change from baseline | -4.1 | -2.5 |
| Difference from placebo | -1.7 | |
| p-value | <0.001 | |
| Monthly headache days across 12 weeks | ||
| Baseline | 9.0 | 8.5 |
| Mean change from baseline | -4.2 | -2.5 |
| Difference from placebo | -1.7 | |
| p-value | <0.001 | |
| Monthly acute medication use days across 12 weeks | ||
| Baseline | 6.9 | 6.5 |
| Mean change from baseline | -3.8 | -2.3 |
| Difference from placebo | -1.4 | |
| p-value | <0.001 | |
| ≥50% MMD responders across 12 weeks | ||
| % Responders | 59 | 29 |
| Odds ratio (95% CI) | 3.55 (2.39, 5.28) | |
| p-value | <0.001 | |
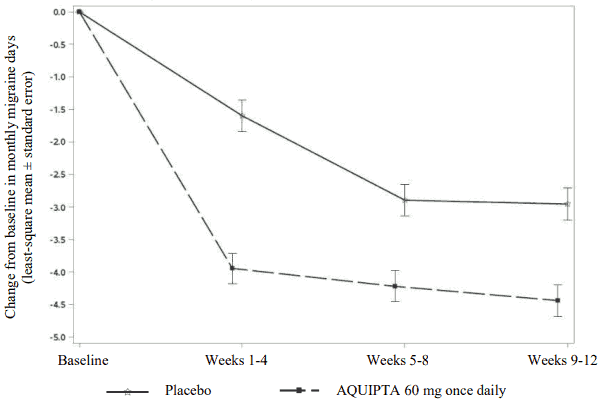
Figure 1 shows the mean change from baseline in MMD in ADVANCE. Patients treated with AQUIPTA 60 mg once daily had greater mean decreases from baseline in MMD across the 12-week treatment period compared to patients who received placebo. AQUIPTA 60 mg once daily resulted in significant decreases from baseline in mean monthly migraine days within the first 4-week interval compared to placebo-treated patients.
Figure 1. Change from baseline in monthly migraine days in ADVANCE:
Long-term efficacy
Efficacy was sustained for up to one year in an open-label study in which 546 patients with episodic migraine were randomised to receive AQUIPTA 60 mg once daily. 68% (373/546) of patients completed the treatment period. The reduction in the least-squares mean number of monthly migraine days in the first month (weeks 1-4) was -3.8 days and improved to a least-squares mean reduction of -5.2 days in the last month (weeks 49-52). Approximately 84%, 70%, and 48% of patients reported ≥50%, ≥75%, and 100% reduction in monthly migraine days at weeks 49-52, respectively.
Chronic migraine
Atogepant was evaluated for the prophylaxis of chronic migraine (15 or more headache days per month with at least 8 migraine days) in a randomised, multicentre, double-blind, placebo-controlled study (PROGRESS). Patients were randomised to AQUIPTA 60 mg (N=262) or placebo (N=259) once daily for 12 weeks. A subset of patients (11%) was allowed to use one concomitant migraine prophylaxis medicinal product (e.g., amitriptyline, propranolol, topiramate). Patients were allowed to use acute headache treatments (i.e., triptans, ergotamine derivatives, NSAIDs, paracetamol and opioids) as needed. Patients with acute medication overuse and medication overuse headache also were enrolled. The use of a concomitant medicinal product that acts on the CGRP pathway was not permitted for either acute or preventive treatment of migraine.
A total of 463 (89%) patients completed the 12-week double-blind study. Patients had a mean age of 42 years (range: 18 to 74 years), 3% were 65 years or older, 87% were female, and 59% were white. The mean migraine frequency at baseline was approximately 19 migraine days per month and was similar across treatment groups.
The primary efficacy endpoint was the change from baseline in mean MMD across the 12-week treatment period. Secondary endpoints controlled for multiplicity included the change from baseline in mean monthly headache days, the change from baseline in mean monthly acute medication use days, the proportion of patients achieving at least a 50% reduction from baseline in mean MMD (3-month average), and several patient-reported outcome measures assessing functioning. Statistically significant findings were demonstrated for AQUIPTA versus placebo for the primary and secondary efficacy endpoints for PROGRESS, as summarized in Table 4.
Table 4. Efficacy endpoints in PROGRESS:
| AQUIPTA 60 mg N=257 | Placebo N=249 | |
|---|---|---|
| Monthly migraine days (MMD) across 12 weeks | ||
| Baseline | 19.2 | 19.0 |
| Mean change from baseline | -6.8 | -5.1 |
| Difference from placebo | -1.7 | |
| p-value | 0.002 | |
| Monthly headache days across 12 weeks | ||
| Baseline | 21.5 | 21.4 |
| Mean change from baseline | -6.9 | -5.2 |
| Difference from placebo | -1.7 | |
| p-value | 0.002 | |
| Monthly acute medication use days across 12 weeks | ||
| Baseline | 15.5 | 15.3 |
| Mean change from baseline | -6.2 | -4.1 |
| Difference from placebo | -2.1 | |
| p-value | 0.002 | |
| ≥50% MMD responders across 12 weeks | ||
| % Responders | 40 | 27 |
| Odds ratio (95% CI) | 1.90 (1.29, 2.79) | |
| p-value | 0.002 | |
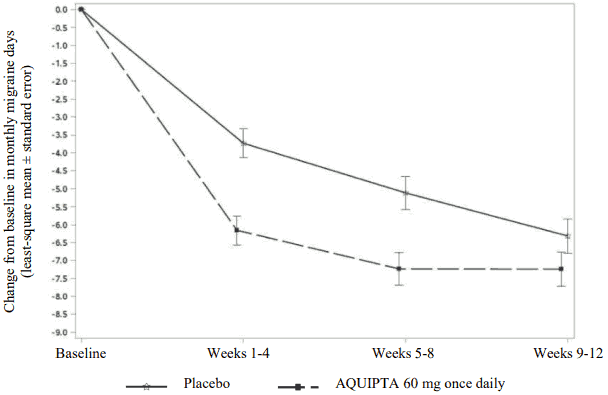
Figure 2 shows the mean change from baseline in MMD in PROGRESS. Patients treated with AQUIPTA 60 mg once daily had a greater mean decrease from baseline in MMD across the 12-week treatment period compared to patients who received placebo.
Figure 2. Change from baseline in monthly migraine days in PROGRESS:
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with AQUIPTA in one or more subsets of the paediatric population in prophylaxis of migraine headaches (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following oral administration, atogepant is absorbed with peak plasma concentrations at approximately 1 to 2 hours. Following once daily dosing, atogepant displays dose-proportional pharmacokinetics up to 170 mg (approximately 3 times the highest recommended dose), with no accumulation.
Effect of food
When atogepant was administered with a high-fat meal, AUC and Cmax were reduced by approximately 18% and 22%, respectively, with no effect on median time to maximum atogepant plasma concentration. Atogepant was administered without regard to food in clinical efficacy studies.
Distribution
Plasma protein binding of atogepant was not concentration-dependent in the range of 0.1 to 10 µM; the unbound fraction of atogepant was approximately 4.7% in human plasma. The mean apparent volume of distribution of atogepant (Vz/F) after oral administration is approximately 292 L.
Biotransformation
Atogepant is eliminated mainly through metabolism, primarily by CYP3A4. The parent compound (atogepant), and a glucuronide conjugate metabolite (M23) were the most prevalent circulating components in human plasma.
CYP3A4 inducers
Co-administration of atogepant with steady state rifampicin, a strong CYP3A4 inducer, resulted in a significant decrease in exposure (Cmax by 30% and AUC by 60%) of atogepant in healthy subjects.
Co-administration of atogepant with steady-state topiramate, a mild CYP3A4 inducer, resulted in a decrease in exposure (Cmax by 24% and AUC by 25%) of atogepant.
In vitro, atogepant is not an inhibitor for CYP3A4, 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, MAO-A, or UGT1A1 at clinically relevant concentrations. Atogepant also is not an inducer of CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
Elimination
The elimination half-life of atogepant is approximately 11 hours. The mean apparent oral clearance (CL/F) of atogepant is approximately 19 L/h. Following single oral dose of 50 mg 14C-atogepant to healthy male subjects, 42% and 5% of the dose was recovered as unchanged atogepant in faeces and urine, respectively.
Transporters
Atogepant is a substrate of P-gp, BCRP, OATP1B1, OATP1B3, and OAT1. Dose adjustment for concomitant use with strong inhibitors of OATP is recommended based on a clinical interaction study with a strong OATP inhibitor. Atogepant is not a substrate of OAT3, OCT2, or MATE1.
Atogepant is not an inhibitor of P-gp, BCRP, OAT1, OAT3, NTCP, BSEP, MRP3, or MRP4 at clinically relevant concentrations. Atogepant is a weak inhibitor of OATP1B1, OATP1B3, OCT1, and MATE1, but no clinically relevant interactions are expected.
Special populations
Renal impairment
The renal route of elimination plays a minor role in the clearance of atogepant. Based on population pharmacokinetic analysis, there is no significant difference in the pharmacokinetics of atogepant in patients with mild or moderate renal impairment (CLcr 30-89 mL/min) relative to those with normal renal function (CLcr ≥90 mL/min). As patients with severe renal impairment or end-stage renal disease (ESRD; CLcr <30 mL/min) have not been studied, use of atogepant 10 mg is recommended in those patients.
Hepatic impairment
In patients with pre-existing mild (Child-Pugh Class A), moderate (Child-Pugh Class B), or severe hepatic impairment (Child-Pugh Class C), total atogepant exposure was increased by 24%, 15% and 38%, respectively. However, unbound atogepant exposure was approximately 3-fold higher in patients with severe hepatic impairment. The use of AQUIPTA in patients with severe hepatic impairment should be avoided.
Other special populations
Based on a population pharmacokinetic analysis, sex, race, and body weight did not have a significant effect on the pharmacokinetics (Cmax and AUC) of atogepant. Therefore, no dose adjustments are warranted based on these factors.
5.3. Preclinical safety data
Notwithstanding marked interspecies differences in CGRP-receptor affinity of atogepant, non-clinical data reveal no special hazard for atogepant in humans based on conventional studies of safety pharmacology, repeat dose toxicity, genotoxicity, phototoxicity or carcinogenic potential.
Impairment of fertility
Oral administration of atogepant to male and female rats prior to and during mating and continuing in females to gestation day 7 resulted in no adverse effects on fertility or reproductive performance. Plasma exposures (AUC) are up to approximately 15 times that in humans at the maximum recommended human dose (MRHD).
Reproductive and developmental toxicology
Oral administration of atogepant to pregnant rats and rabbits during the period of organogenesis resulted in decreased foetal body weight in rats and an increased incidence of foetal visceral and skeletal variations at doses associated with minimal maternal toxicity. At the no-effect dose for adverse effects on embryofoetal development, plasma exposure (AUC) was approximately 4 times in rats and 3 times in rabbits that in humans at the MRHD of 60 mg/day.
Oral administration of atogepant to rats throughout gestation and lactation resulted in non-adverse significant decreased pup body weight which persisted into adulthood. Plasma exposure (AUC) at the no-effect dose for pre- and postnatal development were approximately 5-times that in humans at the MRHD. In lactating rats, oral dosing with atogepant resulted in levels of atogepant in milk approximately 2-fold higher than those in maternal plasma.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.