BALVERSA Film-coated tablet Ref.[111498] Active ingredients: Erdafitinib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein kinase inhibitors
ATC code: L01EN01
Mechanism of action
Erdafitinib is a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor.
Pharmacodynamic effects
Serum phosphate
Erdafitinib increases serum phosphate concentration, a secondary effect of FGFR inhibition (see sections 4.2 and 4.8).
Clinical efficacy
The efficacy of Balversa was evaluated in BLC3001 Study Cohort 1, a Phase 3, randomised, open-label, multicentre study to evaluate the overall survival (OS) of erdafitinib versus chemotherapy (docetaxel or vinflunine) in patients with advanced (unresectable or metastatic) urothelial cancer harbouring selected FGFR alterations, who have progressed after 1 or 2 prior treatments, at least 1 of which includes a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor (anti-PD-(L)1) used in the locally advanced unresectable or metastatic treatment setting.
Patients who received neoadjuvant or adjuvant chemotherapy or immunotherapy and showed disease progression within 12 months of the last dose are considered to have received systemic therapy in the metastatic setting. Patients with uncontrolled cardiovascular disease within the preceding 3 months or with grade 2 or higher (≥481 ms) QTc prolongation and impaired wound healing were excluded from the study, as well as patients with central serous retinopathy or retinal pigment epithelial detachment of any grade.
Main efficacy data are based on 266 patients who received prior anti-PD-(L)1 treatment and were randomised to erdafitinib (8 mg with individualised up-titration to 9 mg if the serum phosphate level is <9.0 mg/dL, and there was no drug-related toxicity) versus chemotherapy (docetaxel 75 mg/m² once every 3 weeks or vinflunine 320 mg/m² once every 3 weeks).
In the study, eligible patients were required to have at least 1 of the following FGFR fusions: FGFR2-BICC1, FGFR2-CASP7, FGFR3-TACC3, FGFR3-BAIAP2L1; or 1 of the following FGFR3 gene mutations: R248C, S249C, G370C, Y373C. Molecular eligibility was determined using central (74.6%) or local (25.4%) FGFR results. Tumour samples were tested for FGFR genetic alterations with the Qiagen Therascreen FGFR RGQ RT-PCR Kit at the central laboratory. Local historical test on tumour or blood samples were based on local next generation sequencing (NGS) tests. Among the limited number of patients enrolled by local tests who had tumour samples available for confirmation testing, a 75.6% agreement was observed when tested using the central test. In the study cohort, 99.2% of patients had FGFR genetic alterations (2 patients did not have FGFR alterations: 80.8% of patients had FGFR3 mutations, 16.5% of patients had FGFR3 fusions, and 1.9% of patients had both FGFR3 mutations and fusions). No patients were observed with FGFR2 alterations in this study cohort. A tumour harbouring susceptible FGFR3 genetic alterations is a tumour with at least 1 of the following FGFR fusions: FGFR3-TACC3, FGFR3-BAIAP2L1; or 1 of the following FGFR3 gene mutations: R248C, S249C, G370C, Y373C. All patients in the study cohort with FGFR alterations had at least 1 FGFR3 alteration. FGFR3-S249C was the most prevalent alteration (46.6%) followed by FGFR3-Y373C (16.9%), and FGFR3-TACC3 fusion (9.8%).
The demographic characteristics were balanced across the erdafitinib and chemotherapy treatment groups. The median age at full-study screening was 67 years (range: 32 to 86 years). The majority of patients were 65 years or older: 19.9% 65 to 69 years; 19.9% 70 to 74 years; 21.1% 75 years or older. The majority of patients were male (71.4%), white (54.1%), and from Europe (60.9%).
All patients had transitional cell carcinoma, with a small percentage (5.3%) of patients having minor components (<50% overall) of variant histology. The primary tumour location was the upper tract for 33.5% of patients and lower tract for 66.5%. Patients had baseline ECOG scores of 0 (42.9%), 1 (47.7%), or 2 (9.4%). All patients received at least 1 prior line of anti-cancer therapy and must have included an anti-PD-(L)-1. The most frequently received anti-PD-(L)1 therapies, included pembrolizumab (35.3%), avelumab (22.2%) and atezolizumab (19.5%). Prior treatment with chemotherapy was not required, however, the majority of patients (89.1%) received at least one line of prior chemotherapy. Almost all patients received platinum-based chemotherapy (89.7% in erdafitinib group, 85.4% in chemotherapy group): most frequently cisplatin (55.9% in erdafitinib group, 45.4% in chemotherapy group) followed by carboplatin (27.2% in erdafitinib group, 31.5% in chemotherapy group).
The primary efficacy endpoint was Overall Survival. Assessment of radiographic response was performed by investigators according to RECIST (Response Evaluation Criteria in Solid Tumours Version 1.1) until disease progression, intolerable toxicity, withdrawal of consent, or decision by the investigator to discontinue treatment, or the end of the study, whichever occurred first. Progression-Free Survival (PFS), Objective Response Rate (ORR) and Duration of Response were included as secondary efficacy endpoints.
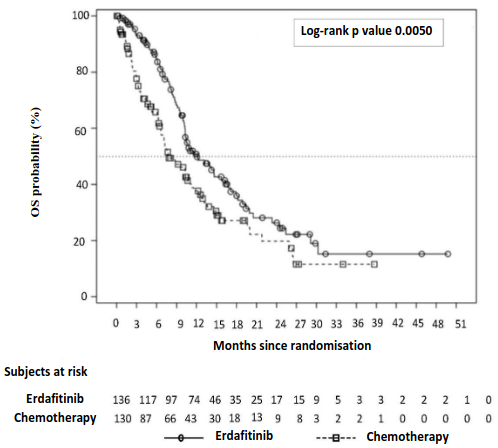
Treatment with erdafitinib showed a statistically significant improvement in OS for patients treated with erdafitinib, with erdafitinib prolonging OS compared to treatment with chemotherapy (median OS of 12.1 vs 7.8 months) (see Table 7).
Efficacy results are summarised in Table 7.
Table 7. Overview of Efficacy Results for Study BLC3001 Cohort 1:
| Erdafitinib (N=136) | Chemotherapy (N=130) | |
|---|---|---|
| Overall Survival (OS) | ||
| Number of events (%) | 77 (56.6%) | 78 (60.0%) |
| Median, months (95% CI) | 12.06 (10.28, 16.36) | 7.79 (6.54, 11.07) |
| HR (95% CI) | 0.64 (0.44, 0.93)a | |
| P-value | 0.0050 | |
| Επιβίωση χωρίς εξέλιξη της νόσου (PFS) | ||
| Number of events (%) | 101 (74.3%) | 90 (69.2%) |
| Median, months (95% CI) | 5.55 (4.40, 5.65) | 2.73 (1.81, 3.68) |
| HR (95% CI) | 0.58 (0.41, 0.82)a | |
| P-value | 0.0002 | |
| Objective response rate (ORR), confirmed | ||
| ORR (CR + PR) | 48 (35.3%) | 11 (8.5%) |
| Duration of response (DoR), investigator assessed, confirmed | ||
| Median, months (95% CI) | 5.55 (4.17, 8.31) | 5.75 (4.86, 7.16) |
All p-values reported are 2-sided.
a Repeated confidence intervals are provided.
The Kaplan-Meier OS curve for the two treatment arms is presented in Figure 1.
Figure 1. Kaplan-Meier Plot of Overall Survival – Unstratified Analysis (BLC3001 Study Cohort 1):
Elderly patients
In the clinical study of Balversa, 60.9% of patients were 65 years and older (39.8% were 65-<75 years old and 21.1% of patients were 75 years and older). No overall difference in efficacy was observed between elderly and younger adult patients.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with erdafitinib in all subsets of the paediatric population in urothelial carcinoma (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Following single and repeat once daily dosing, erdafitinib exposure (maximum observed plasma concentration [Cmax] and area under the plasma concentration time curve [AUC]) increased in a dose-proportional manner across the dose range of 0.5 to 12 mg. Steady state was achieved after 2 weeks with once daily dosing and the mean accumulation ratio was 4-fold in patients with cancer. Following administration of 8 mg once daily, the proposed starting dose, mean (coefficient of variation [CV%]) erdafitinib steady-state Cmax, AUCτ, and minimum observed plasma concentration (Cmin) were 1399 ng/mL (50.8%), 29268 ng.h/mL (59.9%), and 936 ng/mL (64.9%) in patients with cancer. Daily fluctuations in erdafitinib plasma concentrations were low, with a mean (CV%) peak-to- trough ratio of 1.47 (23%) at steady state upon daily dosing.
Absorption
After single dose oral administration, median time to achieve peak plasma concentration (tmax) was 2.5 hours (range: 2 to 6 hours) in healthy volunteers and oral absorption is near complete.
Effect of food
Administration of erdafitinib to healthy volunteers under fasting conditions and with a high-fat meal did not result in clinically relevant changes in Cmax and AUC. The mean AUC∞ and Cmax decreased by 6% and 14%, respectively, when erdafitinib is co-administered with a high-fat meal. Median time to reach tmax was delayed about 1.5 hours with food (see section 4.2).
Distribution
The mean apparent volume of distribution of erdafitinib in patients with cancer was 0.411 L/kg. Erdafitinib was 99.7% bound to human plasma proteins, preferentially to α1 acid glycoprotein.
Biotransformation
Metabolism is the main route of elimination for erdafitinib. Erdafitinib is primarily metabolised in human by CYP2C9 and CYP3A4 to form the O demethylated major metabolite. The contribution of CYP2C9 and CYP3A4 in the total clearance of erdafitinib is estimated to be 39% and 20%, respectively. Unchanged erdafitinib was the major drug-related moiety in plasma, there were no circulating metabolites.
Elimination
Mean total apparent clearance (CL/F) of erdafitinib was 0.362 L/h in patients with cancer. The mean effective half-life of erdafitinib in patients with cancer was 58.9 hours. Up to 16 days following a single oral administration of radiolabelled [14C]-erdafitinib, 69% of the dose was recovered in faeces (14-21% as unchanged erdafitinib) and 19% in urine (13% as unchanged erdafitinib) in healthy volunteers.
Special populations
No clinically meaningful differences in the pharmacokinetics of erdafitinib were observed based on age (21-92 years), sex, race (White, Hispanic or Asian), body weight (36-166 kg), mild or moderate renal impairment and mild or moderate hepatic impairment.
Paediatric population
Pharmacokinetics of erdafitinib has not been studied in paediatric patients.
Renal impairment
No clinically meaningful differences in the pharmacokinetics of erdafitinib were observed between subjects with normal renal function (absolute GFR-MDRD [absolute glomerular filtration rate modification of diet in renal disease] ≥90 mL/min), and subjects with mild (absolute GFR‑MDRD 60 to 89 mL/min) and moderate renal impairment (absolute GFR‑MDRD 30 to 59 mL/min) based on population PK analysis. No information is available for subjects with severe renal impairment (absolute GFR‑MDRD less than 30 mL/min) or renal impairment requiring dialysis due to scarcity of PK data (n=7, 0.8%).
Hepatic impairment
The pharmacokinetics of erdafitinib was examined in participants with preexisting mild n=8) or moderate n=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in healthy control participants with normal hepatic function (n=8). The total AUC∞ were 82% and 61% in participants with mild and moderate hepatic impairment compared with participants with normal hepatic function, respectively. The total Cmax were 83% and 74% in participants with mild and moderate hepatic impairment compared with participants with normal hepatic function, respectively. The free AUC∞ were 95% and 88% in participants with mild and moderate hepatic impairment compared with participants with normal hepatic function, respectively. The free Cmax were 96% and 105% in participants with mild and moderate hepatic impairment compared with participants with normal hepatic function, respectively. No clinically meaningful differences in the pharmacokinetics of erdafitinib were observed in subjects with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment and subjects with normal hepatic function. The pharmacokinetics of erdafitinib in subjects with severe hepatic impairment is unknown due to limited data.
Drug interactions
Effect of P-gp inhibitors on erdafitinib
Erdafitinib is a substrate for P-gp. P-gp inhibitors are not expected to affect the PK of erdafitinib in a clinically relevant manner.
Effect of acid lowering agents on erdafitinib
Erdafitinib has adequate solubility across the pH range of 1 to 7.4. Acid lowering agents (e.g., antacids, H2-antagonists, or proton pump inhibitors) are not expected to affect the bioavailability of erdafitinib.
Effect of Sevelamer on erdafitinib
No clinically meaningful differences in the pharmacokinetics of erdafitinib were observed in patients taking sevelamer.
5.3. Preclinical safety data
Repeat-dose toxicity
The main toxicological findings following repeat-dose administration of erdafitinib in both rats and dogs were related to the pharmacological activity of erdafitinib as an irreversible inhibitor of FGFR, including increased inorganic phosphorus and calcium in plasma, ectopic mineralisation in various organs and tissues, lesions in bone/cartilage at erdafitinib exposures lower than the human exposure at the recommended clinical dose. Corneal atrophy (thinning of the corneal epithelium) was seen in rats and lacrimal gland atrophy, changes to haircoat and nails as well as dental changes after 3 months of treatment was seen in rats and dogs. Disturbance of phosphate homeostasis was observed in rats and dogs at exposures less than the human exposures at all doses studied.
Soft tissue mineralisations (except for the aorta mineralisation in dogs) and chondroid dysplasia in rats and dogs and mammary gland atrophy in rats were partially to fully recovered at the end of a 4-week drug-free recovery period.
Erdafitinib is an intrinsic human ether-à-go-go-related gene (hERG) blocker with a proarrhythmic liability which translated into a prolonged repolarisation (corrected QT interval) after intravenous dosing in the anaesthetised dog and guinea pig, and after oral dosing in the conscious dog. The no effect level represents a safety margin of 2.4 relative to the clinical steady-state free maximum plasma concentration (Cmax,u) for a 9 mg once daily dose.
Carcinogenicity and mutagenicity
Long-term animal studies have not been conducted to evaluate the carcinogenic potential of erdafitinib. Erdafitinib was considered not genotoxic in the standard panel of good laboratory practice (GLP) genotoxicity assays.
Reproductive toxicology
Erdafitinib was teratogenic and embryotoxic in rats at exposures less than the human exposures. Foetal toxicity was characterised by hand/foot defects and malformations of some major blood vessels, such as the aorta (see sections 4.4 and 4.6).
Fertility
Dedicated animal fertility studies have not been conducted with erdafitinib. However, in the 3-month general toxicity study, erdafitinib showed effects on female reproductive organs (necrosis of the corpora lutea) in rats at an exposure approximating the AUC in patients at maximum recommended dose of 9 mg, QD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.