BENLYSTA Powder for concentrate for solution for infusion / Solution for injection Ref.[110232] Active ingredients: Belimumab
Source: FDA, National Drug Code (US) Revision Year: 2024
12. Clinical Pharmacology
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of BENLYSTA or of other belimumab products.
In Trials 2 and 3 (intravenous dosing in adults with SLE), anti-belimumab antibodies were assessed during the respective 52-week and 76-week, placebo-controlled periods and detected in 4 of 563 (0.7%) patients receiving BENLYSTA 10 mg/kg and in 27 of 559 (4.8%) patients receiving BENLYSTA 1 mg/kg. The reported frequency for the group receiving 10 mg/kg may underestimate the actual frequency due to lower assay sensitivity in the presence of high drug concentrations. Neutralizing antibodies were detected in 3 patients receiving BENLYSTA 1 mg/kg. Three patients with anti-belimumab antibodies experienced mild infusion reactions of nausea, erythematous rash, pruritus, eyelid edema, headache, and dyspnea; none of the reactions were life-threatening. In Trial 4 (intravenous dosing in adult Black patients), anti-belimumab antibodies were detected in 2 of 321 (0.6%) patients receiving BENLYSTA 10 mg/kg during the 52-week, placebo-controlled period. In Trial 5 (intravenous dosing in adults with lupus nephritis), there was no formation of anti-belimumab antibodies in 224 patients receiving BENLYSTA 10 mg/kg plus standard therapy during the 104-week, placebo‑controlled period. In Trial 6 (intravenous dosing in pediatric patients with SLE), there was no formation of anti-belimumab antibodies in 53 patients receiving BENLYSTA 10 mg/kg plus standard therapy during the 52-week, placebo‑controlled period. In Trial 7 (subcutaneous dosing in adults with SLE), there was no formation of anti‑belimumab antibodies in 556 patients receiving BENLYSTA 200 mg during the 52-week, placebo-controlled period.
The clinical relevance of the presence of anti-belimumab antibodies is not known.
12.1. Mechanism of Action
BENLYSTA is a BLyS-specific inhibitor that blocks the binding of soluble BLyS, a B-cell survival factor, to its receptors on B cells. BENLYSTA does not bind B cells directly, but by binding BLyS, BENLYSTA inhibits the survival of B cells, including autoreactive B cells, and reduces the differentiation of B cells into immunoglobulin-producing plasma cells.
12.2. Pharmacodynamics
Treatment with BENLYSTA in adult patients significantly reduced circulating CD19+, CD20+, naïve, and activated B cells, and the SLE B‑cell subset at Week 52. Reductions in naïve and the SLE B‑cell subset were observed as early as Week 8 and sustained to Week 52. Memory cells increased initially and slowly declined toward baseline levels by Week 52.
Treatment with BENLYSTA in adult patients led to reductions in IgG and anti-double-stranded DNA antibodies (anti-dsDNA) which were observed as early as Week 8 and sustained through Week 52. In patients with low complement levels at baseline, treatment led to increases in complement C3 and C4 as early as Week 12 and were sustained through Week 52.
The pharmacodynamic response observed in Black patients (Trial 4) was consistent with the previous studies.
In patients with active lupus nephritis (Trial 5), following treatment with BENLYSTA, there was a decrease in serum IgG as early as Week 4, and subsequently there was an increase in serum IgG levels which was associated with decreased proteinuria. Reductions in autoantibodies, increases in complement, and reductions in circulating total B cells and B‑cell subsets observed were consistent with the SLE studies.
In Trial 6 (pediatric dosing), the pharmacodynamic response was consistent with the adult data.
The clinical relevance of above mentioned pharmacodynamic biomarkers has not been established.
12.3. Pharmacokinetics
Intravenous Infusion in Adults
Systemic Lupus Erythematosus
The pharmacokinetic parameters displayed in Table 2 are based on population parameter estimates from 563 adult patients who received BENLYSTA 10 mg/kg.
Table 2. Population Pharmacokinetic Parameters in Adult Patients with SLE after Intravenous Infusion of BENLYSTA 10 mg/kga:
| Pharmacokinetic Parameter | Population Estimates (n=563) |
|---|---|
| Peak concentration (Cmax, mcg/mL) | 313 |
| Area under the curve (AUC0-∞, day•mcg/mL) | 3,083 |
| Distribution half-life (t½, days) | 1.8 |
| Terminal half-life (t½, days) | 19.4 |
| Systemic clearance (CL, mL/day) | 215 |
| Volume of distribution (Vss, L) | 5 |
a Intravenous infusions were administered at 2-week intervals for the first 3 doses and at 4-week intervals thereafter.
Lupus Nephritis: A population pharmacokinetic analysis was conducted in 224 adult patients with lupus nephritis who received belimumab 10 mg/kg intravenously (Days 0, 14, 28, and then every 28 days up to 104 weeks) plus standard therapy [see Clinical Studies (14.2)]. In patients with lupus nephritis, due to additional clearance associated with proteinuria, belimumab exposure was initially lower than observed in SLE studies and lower belimumab exposure was observed in patients with higher proteinuria. When the proteinuria was decreased to approximately ≤1 g/g after treatment, belimumab clearance and exposure were similar to that observed in patients with SLE who received belimumab 10 mg/kg intravenously. The available data do not support a dose adjustment in patients with high proteinuria.
Subcutaneous Injection in Adults
Systemic Lupus Erythematosus
The pharmacokinetic parameters displayed in Table 3 are based on population parameter estimates from 661 subjects after subcutaneous administration of belimumab 200 mg once weekly. The time to reach maximum serum concentration (Cmax) was 2.6 days (Tmax) after administration at steady state. The bioavailability of belimumab was approximately 74%. With weekly subcutaneous administration there were minor fluctuations around the average concentration (Cavg 104 mcg/mL), with Cmin (97 mcg/mL) being only slightly below Cavg.
Table 3. Population Pharmacokinetic Parameters in Adults after Subcutaneous Administration of BENLYSTA:
| Pharmacokinetic Parameter | Population Estimates (n=661) |
|---|---|
| Peak concentration (Cmax, mcg/mL) | 108 |
| Area under the curve (AUC0-∞, day•mcg/mL) | 726 |
| Distribution half-life (t½, days) | 1.1 |
| Terminal half-life (t½, days) | 18.3 |
| Systemic clearance (CL, mL/day) | 204 |
| Volume of distribution (Vss, L) | 5 |
Lupus Nephritis
Based on population pharmacokinetic modeling and simulation of the subcutaneous 400-mg weekly loading dose, the average belimumab concentration during the first 12 weeks was predicted to be 78 mcg/mL, which is similar to the estimated concentration of 89 mcg/mL for intravenous administration. The loading dose of 400 mg weekly provides steady-state concentrations from Week 2 of dosing. The steady-state average concentrations of subcutaneous administration of belimumab 200 mg once weekly in adults with lupus nephritis are predicted to be similar to those observed in adults with lupus nephritis receiving belimumab 10 mg/kg intravenously every 4 weeks.
Specific Population
The following information is based on the population pharmacokinetic analyses of intravenous administration and subcutaneous administration of BENLYSTA.
Age
Age did not significantly influence the pharmacokinetics of belimumab, where the majority of subjects were between 18 and 45 years (70% with intravenous dosing; 74% with subcutaneous dosing).
Geriatric Patients: Limited pharmacokinetic data are available for elderly patients as less than 2% of the subjects included in the pharmacokinetic analysis were 65 years or older [see Use in Specific Populations (8.5)].
Pediatric Patients: The pharmacokinetic parameters of belimumab are based on individual parameter estimates from a population pharmacokinetic analysis of 53 pediatric patients with SLE (Trial 6). Following IV administration of 10 mg/kg on Days 0, 14, and 28, and at 4‑week intervals thereafter, belimumab exposures were similar between pediatric and adult subjects with SLE. Steady-state geometric mean Cmax, Cmin, Cavg, and AUC values were 305, 42, 92 mcg/mL, and 2,569 day•mcg/mL in the 5- to 11-year-old group, and 317, 52, 112 mcg/mL and 3,126 day•mcg/mL in the 12- to 17-year-old group [See Use in Specific Populations (8.4)].
For active lupus nephritis, the pharmacokinetics of belimumab in pediatric patients were estimated based on a population pharmacokinetic model developed from 224 adults with active lupus nephritis and validated using data from 53 pediatric patients with SLE. With IV administration of 10 mg/kg on Days 0, 14 and 28 and at 4-week intervals thereafter, the simulated belimumab exposures for both the 5- to 11-year-old group and the 12- to 17-year-old group were estimated to be comparable to adults with active lupus nephritis [See Use in Specific Populations (8.4)].
Male and Female Patients
Gender did not significantly influence belimumab pharmacokinetics in the largely female trial population (94% with intravenous dosing; 85% with subcutaneous dosing).
Racial Groups
Race did not significantly influence belimumab pharmacokinetics. The racial distribution with intravenous administration was 53% White, 16% Asian, 16% Alaska native/American Indian, and 14% Black in Trials 1, 2, and 3. Trial 4 enrolled only Black patients. The racial distribution with subcutaneous administration (Trial 7) was 61% White, 20% Asian, 11% Black, and 6% Alaska native/American Indian.
Weight
Body weight and body mass index (BMI) had no clinically relevant effect on the pharmacokinetics of belimumab administered subcutaneously in adults. No dose adjustment is recommended based on weight or BMI for subcutaneous administration.
Patients with Renal Impairment
No formal trials were conducted to examine the effects of renal impairment on the pharmacokinetics of belimumab. BENLYSTA was studied in a limited number of adult patients with SLE who had mild (CrCl ≥60 and <90 mL/min), moderate (CrCl ≥30 and <60 mL/min), or severe (CrCl ≥15 and <30 mL/min) renal impairment: 770 patients with mild renal impairment, 261 patients with moderate renal impairment, and 14 patients with severe renal impairment received belimumab intravenously; 121 patients with mild renal impairment and 30 patients with moderate renal impairment received belimumab subcutaneously [See Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
No formal trials were conducted to examine the effects of hepatic impairment on the pharmacokinetics of belimumab. Baseline alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels did not significantly influence belimumab pharmacokinetics [See Use in Specific Populations (8.7)].
Drug Interaction Studies
No formal drug interaction studies have been conducted with BENLYSTA. Concomitant use of mycophenolate, cyclophosphamide, azathioprine, methotrexate, antimalarials, NSAIDs, aspirin, and/or HMG-CoA reductase inhibitors did not significantly influence belimumab pharmacokinetics. Coadministration of steroids and angiotensin-converting enzyme (ACE) inhibitors resulted in an increase of systemic clearance of belimumab that was not clinically significant because the magnitude was well within the range of normal variability of clearance. The effect of belimumab on the pharmacokinetics of other drugs has not been evaluated.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of belimumab.
Effects on male and female fertility have not been directly evaluated in animal studies.
14. Clinical Studies
The safety and effectiveness of BENLYSTA administered intravenously plus standard therapy were evaluated in 4 randomized, double‑blind, placebo‑controlled trials involving 2,581 adult patients with SLE (Trial 1, NCT00071487, Trial 2, NCT00410384, Trial 3, NCT00424476, and Trial 4 NCT01632241), and one trial involving 93 pediatric patients (Trial 6, NCT01649765) with SLE according to the American College of Rheumatology criteria. In these trials, patients with severe active lupus nephritis and severe active CNS lupus were excluded. Patients were on a stable standard therapy SLE treatment regimen comprising any of the following (alone or in combination): corticosteroids, antimalarials, NSAIDs, and immunosuppressives. Use of other biologics and intravenous cyclophosphamide was not permitted.
In addition, the safety and effectiveness of BENLYSTA administered intravenously plus standard therapy was evaluated in a randomized, double‑blind, placebo‑controlled trial in 448 adult patients with active lupus nephritis (Trial 5; NCT01639339).
14.1 Intravenous Administration in Adults with SLE
Trial 1: SLE – BENLYSTA 1 mg/kg, 4 mg/kg, 10 mg/kg – Intravenous
Trial 1 enrolled 449 patients and evaluated doses of 1, 4, and 10 mg/kg BENLYSTA plus standard therapy compared with placebo plus standard therapy over 52 weeks in patients with SLE. Patients had to have a Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) score of >4 at baseline and a history of autoantibodies (anti-nuclear antibody [ANA] and/or anti–double-stranded DNA [anti-dsDNA]), but 28% of the population was autoantibody negative at baseline. The co-primary endpoints were percent change in SELENA-SLEDAI score at Week 24 and time to first flare over 52 weeks. No significant differences between any of the groups receiving BENLYSTA and the group receiving placebo were observed. Exploratory analysis of this trial identified a subgroup of patients (72%) who were autoantibody positive in whom BENLYSTA appeared to offer benefit. The results of this trial informed the design of Trials 2 and 3 and led to the selection of a target population and indication that is limited to autoantibody-positive SLE patients.
Trials 2, 3 and 4: SLE – BENLYSTA 1 mg/kg and 10 mg/kg – Intravenous
Trials 2 and 3 were randomized, double‑blind, placebo‑controlled trials in patients with SLE that were similar in design except duration; Trial 2 (N=819) was 76 weeks' duration and Trial 3 (N=865) was 52 weeks' duration. Patients had active SLE disease with a SELENA‑SLEDAI score ≥6 and positive autoantibody test results at screening. Patients were excluded from the trial if they had ever received treatment with a B‑cell-targeted agent or if they were currently receiving other biologic agents. Intravenous cyclophosphamide was not permitted within the previous 6 months or during the trial. Trial 2 was conducted primarily in North America and Europe. Trial 3 was conducted in South America, Eastern Europe, Asia, and Australia.
Baseline concomitant medications included corticosteroids (Trial 2: 76%, Trial 3: 96%), immunosuppressives (Trial 2: 56%, Trial 3: 42%; including azathioprine, methotrexate, and mycophenolate), and antimalarials (Trial 2: 63%, Trial 3: 67%). Most patients (>70%) were receiving 2 or more classes of SLE medications.
In Trial 2 and Trial 3, more than 50% of patients had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA-SLEDAI were mucocutaneous (82% in both trials), immune (Trial 2: 74%, Trial 3: 85%), and musculoskeletal (Trial 2: 73%, Trial 3: 59%). Less than 16% of patients had some degree of renal activity and less than 7% of patients had activity in the vascular, cardio-respiratory, or CNS systems.
At screening, patients were stratified by disease severity based on their SELENA‑SLEDAI score (≤9 vs. ≥10), proteinuria level (<2 g/24 h vs. ≥2 g/24 h), and race (African or Indigenous-American descent vs. other), and then randomly assigned to receive BENLYSTA 1 mg/kg, BENLYSTA 10 mg/kg, or placebo in addition to standard therapy. The patients were administered trial medication intravenously over a 1‑hour period on Days 0, 14, 28, and then every 28 days for 48 weeks in Trial 3 and for 72 weeks in Trial 2.
The primary efficacy endpoint was a composite endpoint (SLE Responder Index-4 or SRI-4) that defined response as meeting each of the following criteria at Week 52 compared with baseline:
- ≥4‑point reduction in the SELENA‑SLEDAI score, and
- no new British Isles Lupus Assessment Group (BILAG) A organ domain score or 2 new BILAG B organ domain scores, and
- no worsening (<0.30‑point increase) in Physician’s Global Assessment (PGA) score.
The SRI uses the SELENA‑SLEDAI score as an objective measure of reduction in global disease activity; the BILAG index to ensure no significant worsening in any specific organ system; and the PGA to ensure that improvements in disease activity are not accompanied by worsening of the patient’s condition overall.
In both Trials 2 and 3, the proportion of patients with SLE achieving an SRI-4 response, as defined for the primary endpoint, was significantly higher in the group receiving BENLYSTA 10 mg/kg plus standard therapy than in the group receiving placebo plus standard therapy. The effect on the SRI-4 was not consistently significantly different for patients receiving BENLYSTA 1 mg/kg plus standard therapy relative to placebo plus standard therapy in both trials. The 1-mg/kg dose is not recommended. The trends in comparisons between the treatment groups for the rates of response for the individual components of the endpoint were generally consistent with that of the SRI-4 (Table 4). At Week 76 in Trial 2, the SRI-4 response rate with BENLYSTA 10 mg/kg was not significantly different from that of placebo (39% and 32%, respectively).
Table 4. Clinical Response Rate in Patients with SLE after 52 Weeks of Treatment:
| Trial 2 | Trial 3 | |||||
|---|---|---|---|---|---|---|
| Response | Placebo + Standard Therapy (n=275) | BENLYSTA 1 mg/kg + Standard Therapy (n=271) | BENLYSTA 10 mg/kg + Standard Therapy (n=273) | Placebo + Standard Therapy (n=287) | BENLYSTA 1 mg/kg + Standard Therapy (n=288) | BENLYSTA 10 mg/kg + Standard Therapy (n=290) |
| SLE Responder Index-4 (SRI-4)b | 34% | 41% P = 0.104 | 43% P = 0.021 | 44% | 51% P = 0.013 | 58% P <0.001 |
| Odds Ratio (95% CI) vs. placebo | 1.3 (0.9, 1.9) | 1.5 (1.1, 2.2) | 1.6 (1.1, 2.2) | 1.8 (1.3, 2.6) | ||
| Components of SLE Responder Index-4 (SRI-4) | ||||||
| Percent of patients with reduction in SELENA- SLEDAI ≥4 | 36% | 43% | 47% | 46% | 53% | 58% |
| Percent of patients with no worsening by BILAG index | 65% | 75% | 69% | 73% | 79% | 81% |
| Percent of patients with no worsening by PGA | 63% | 73% | 69% | 69% | 79% | 80% |
a The 1-mg/kg dose is not recommended.
b Patients dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. In both trials, a higher proportion of placebo patients were considered as failures for this reason compared with the groups receiving BENLYSTA.
The reduction in disease activity seen in the SRI-4 was related primarily to improvement in the most commonly involved organ systems; namely, mucocutaneous, musculoskeletal, and immune.
Effect in Black/African-American Patients: In Trials 2 and 3, exploratory sub-group analyses of SRI-4 response rate in Black patients (n=148) were performed. The SRI-4 response rate in Black patients in groups receiving BENLYSTA plus standard therapy was less than that in the group receiving placebo plus standard therapy (22/50 or 44% for placebo, 15/48 or 31% for BENLYSTA 1 mg/kg, and 18/50 or 36% for BENLYSTA 10 mg/kg).
Trial 4 was a 2:1 randomized, placebo-controlled trial in Black patients with SLE (N=448) conducted in North America, South America, Europe, and Africa (same study design as Trials 2 and 3 with exceptions of patients having a baseline SELENA-SLEDAI score of ≥8 and using the modified SLEDAI-2K scoring for proteinuria). The population had a mean age of 39 years (range: 18 to 71) and 97% were female. The proportion of Black patients achieving an SRI-S2K response at Week 52 (primary endpoint), and the individual components of the endpoint, were higher in the group receiving BENLYSTA 10 mg/kg plus standard therapy relative to the group receiving placebo plus standard therapy. However, the treatment difference was not statistically significant (Table 5).
Table 5. Clinical Response Rate in Black Patients with SLE after 52 Weeks of Treatment (Trial 4):
| Responsea | Placebo + Standard Therapy (n=149) | BENLYSTA 10 mg/kg + Standard Therapy (n=298) |
|---|---|---|
| SLE Responder Index (SRI-S2K)b | 42% | 49% |
| Odds Ratio (95% CI) | 1.4 (0.9, 2.1) P = 0.107 | |
| Components of SLE Responder Index (SRI-S2K) | ||
| Percent of patients with reduction in SELENA‑SLEDAI-S2K ≥4 | 42% | 50% |
| Odds Ratio (95% CI) | 1.5 (1.0, 2.2) | |
| Percent of patients with no worsening by BILAG index | 62% | 68% |
| Odds Ratio (95% CI) | 1.2 (0.8, 1.9) | |
| Percent of patients with no worsening by PGA | 64% | 70% |
| Odds Ratio (95% CI) | 1.3 (0.8, 1.9) | |
a Analyses excluded any subject missing a baseline assessment for any of the components (1 for belimumab).
b Patients dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. A higher proportion of patients receiving placebo were considered as failures for this reason compared with the group receiving BENLYSTA.
Effect on Concomitant Steroid Treatment: In Trial 2 and Trial 3, 46% and 69% of patients, respectively, were receiving prednisone at doses >7.5 mg/day at baseline. The proportion of patients able to reduce their average prednisone dose by at least 25% to ≤7.5 mg/day during Weeks 40 through 52 was not consistently significantly different for BENLYSTA plus standard therapy relative to placebo plus standard therapy in both trials. In Trial 2, 17% of patients receiving BENLYSTA 10 mg/kg plus standard therapy and 19% of patients receiving BENLYSTA 1 mg/kg plus standard therapy achieved this level of steroid reduction compared with 13% of patients receiving placebo plus standard therapy. In Trial 3, 19%, 21%, and 12% of patients receiving BENLYSTA 10 mg/kg, BENLYSTA 1 mg/kg, and placebo, respectively, plus standard therapy achieved this level of steroid reduction.
Effect on Severe SLE Flares: The probability of experiencing a severe SLE flare, as defined by a modification of the SELENA Trial flare criteria, which excluded severe flares triggered only by an increase of the SELENA-SLEDAI score to >12, was calculated for both Trials 2 and 3. The proportion of patients having at least 1 severe flare over 52 weeks was not consistently significantly different for BENLYSTA plus standard therapy relative to placebo plus standard therapy in both trials. In Trial 2, 18% of patients receiving BENLYSTA 10 mg/kg plus standard therapy and 16% of patients receiving BENLYSTA 1 mg/kg plus standard therapy had a severe flare compared with 24% of patients receiving placebo plus standard therapy. In Trial 3, 14%, 18%, and 23% of patients receiving BENLYSTA 10 mg/kg, BENLYSTA 1 mg/kg and placebo, respectively, plus standard therapy had a severe flare.
14.2 Intravenous Administration in Adults with Lupus Nephritis
Trial 5: Lupus Nephritis – BENLYSTA 10 mg/kg – Intravenous
The safety and effectiveness of BENLYSTA 10 mg/kg administered intravenously over 1 hour on Days 0, 14, 28, and then every 28 days plus standard therapy were evaluated in a 104-week, randomized, double‑blind, placebo‑controlled trial in 448 patients with active proliferative and/or membranous lupus nephritis (Trial 5). The patients had a clinical diagnosis of SLE according to American College of Rheumatology classification criteria; biopsy-proven lupus nephritis Class III, IV, and/or V; and had active renal disease at screening requiring standard therapy: corticosteroids with 1) mycophenolate for induction followed by mycophenolate for maintenance, or 2) cyclophosphamide for induction followed by azathioprine for maintenance. This trial was conducted in Asia, North America, South America, and Europe. The mean age of patients was 33 years (range: 18 to 77); the majority (88%) were female.
The primary efficacy endpoint was Primary Efficacy Renal Response (PERR) at Week 104, defined as a response at Week 100 confirmed by a repeat measurement at Week 104 of the following parameters: urine protein:creatinine ratio (uPCR) ≤0.7 g/g and estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73 m² or no decrease in eGFR of >20% from pre-flare value.
The major secondary endpoints included:
- Complete Renal Response (CRR) defined as a response at Week 100 confirmed by a repeat measurement at Week 104 of the following parameters: uPCR <0.5 g/g and eGFR ≥90 mL/min/1.73 m² or no decrease in eGFR of >10% from pre-flare value.
- PERR at Week 52.
- Time to renal-related event or death (renal-related event defined as first event of end-stage renal disease, doubling of serum creatinine, renal worsening [defined by quantified increase in proteinuria and/or impaired renal function], or receipt of renal disease-related prohibited therapy due to inadequate lupus nephritis control or renal flare management).
The proportion of patients achieving PERR at Week 104 was significantly higher in patients receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 6). The major secondary endpoints also showed significant improvement with BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 6 and Table 7).
Table 6. Efficacy Results in Adults with Lupus Nephritis (Trial 5):
| Efficacy Endpointa | Placebo + Standard Therapy n=223 | BENLYSTA + Standard Therapy n=223 | Odds Ratio (OR) vs. Placebo (95% CI) |
|---|---|---|---|
| Primary Efficacy Renal Response (PERR) at Week 104b,c | |||
| Responders | 32% | 43% | 1.6 (1.0, 2.3) P = 0.031 |
| Components of PERR | |||
| Urine protein:creatinine ratio ≤0.7 g/g | 34% | 44% | 1.5 (1.0, 2.3) |
| eGFR ≥60 mL/min/1.73 m² or no decrease in eGFR from pre-flare value of >20% | 50% | 57% | 1.3 (0.9, 1.9) |
| Complete Renal Response (CRR) at Week 104b,c | |||
| Responders | 20% | 30% | 1.7 (1.1, 2.7) P = 0.017 |
| Components of CRR | |||
| Urine protein:creatinine ratio <0.5 g/g | 29% | 39% | 1.6 (1.1, 2.4) |
| eGFR ≥90 mL/min/1.73 m² or no decrease in eGFR from pre-flare value of >10% | 40% | 47% | 1.3 (0.9, 2.0) |
| PERR at Week 52b | |||
| Responders | 35% | 47% | 1.6 (1.1, 2.4) P = 0.025 |
eGFR = Estimated glomerular filtration rate.
a PERR at Week 104 was the primary efficacy analysis; CRR at Week 104 and PERR at Week 52 were included in pre-specified testing hierarchy.
b In order to be considered a responder, steroid treatment had to be reduced to ≤10 mg/day from Week 24. Patients who discontinued treatment early, received prohibited medication or increases in background standard therapy, or withdrew from the study were considered non-responders. Prohibited medications and increases in background standard therapy were defined as: 1) use of corticosteroids above that allowed by protocol; 2) additional immunosuppressive agents (except topicals) beyond their induction/maintenance regimens; 3) angiotensin converting enzyme inhibitors (ACE) inhibitors, angiotensin II receptor blockers (ARBs), or antimalarials initiated after Week 24; 4) exceeding protocol-permitted doses for standard therapy (cyclophosphamide, azathioprine, mycophenolate); or 5) other biologics, IV immunoglobulin, or plasmapheresis.
c The percentage of patients who did not take prohibited medications or have an increase in background standard therapy at Week 104 was 83% for BENLYSTA and 74% for placebo.
In descriptive subgroup analyses, the PERR and CRR rates were examined by induction therapy (mycophenolate or cyclophosphamide), biopsy class (Class III or IV, Class III + V or Class IV + V, or Class V), and uPCR levels at baseline (<3 g/g or ≥3 g/g; post-hoc analysis) (Figure 1).
Figure 1. Odds Ratio of PERR and CRR at Week 104 across Subgroupsa,b (Trial 5):
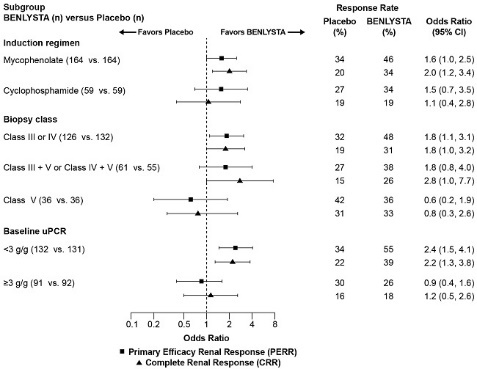
a Class III = Focal proliferative lupus nephritis; Class IV = Diffuse proliferative lupus nephritis; Class V = Membranous lupus nephritis; Class III + V = Mixed membranous-focal proliferative lupus nephritis; Class IV + V = Mixed membranous-diffuse proliferative lupus nephritis.
b Baseline urine protein:creatinine ratio (uPCR) was a post-hoc analysis.
The proportion of responders for PERR by visit through Week 104 is shown in Figure 2.
Figure 2. Primary Efficacy Renal Response (PERR) in Adults with Lupus Nephritis (+/- Standard Error) by Visita (Trial 5):
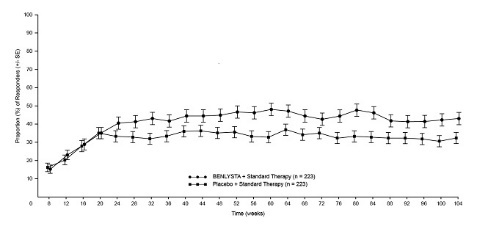
a Analysis is descriptive; bars represent standard error. The same patients may not have responded at each timepoint.
In Trial 5, subjects who received BENLYSTA were significantly less likely to experience a renal-related event or death compared with placebo (Table 7).
Table 7. Time to Renal-Related Event or Death in Adults with Lupus Nephritis (Trial 5):
| Efficacy Endpoint | Placebo + Standard Therapy n=223 | BENLYSTA + Standard Therapy n=223 | Hazard Ratio (HR) vs. Placebo (95% CI) |
|---|---|---|---|
| Time to Renal-Related Event or Deatha | |||
| Percentage of patients with eventb Number of patients with event | 28% 63 | 16% 35 | |
| Time to eventc | 0.5 (0.3, 0.8) P = 0.001 | ||
| Components of endpointd Percentage of patients with event | |||
| End-stage renal disease (ESRD) | 0.4% | 1% | |
| Doubling of serum creatinine from baseline | 4% | 1% | |
| Renal worseninge | 18% | 8% | |
| Renal-related treatment failuref | 16% | 9% | |
| Death | 1% | 0.4% |
a Time to renal-related event or death included in pre-specified testing hierarchy.
b When excluding deaths from analysis (1 for BENLYSTA; 2 for placebo), the percentage of patients with a renal-related event was 15% for BENLYSTA compared with 27% for placebo (HR = 0.5; 95% CI: 0.3, 0.8).
c Subjects who discontinued treatment, were withdrawn from the study, lost to follow-up, or had a treatment failure not related to renal disease were censored on the date of the event. Subjects who completed the 104-week treatment period were censored at the Week 104 visit. Time to event was defined as (event date minus treatment start date plus 1 day).
d Patients could have had more than one event; the first event contributed to the overall endpoint.
e Renal worsening was prospectively defined as the development of increased proteinuria and/or impaired renal function defined as: 1) Increased proteinuria (using spot urine): a reproducible increase in 24-hour urine protein levels to >1 g/g if the baseline value was <0.2 g/g or >2 g/g if the baseline value was between 0.2 g/g and 1 g/g or more than twice the value at baseline if the baseline value was >1 g/g; and 2) Impaired renal function: a reproducible decrease in eGFR of >20% accompanied by at least 1 of the following: proteinuria (>1 g/g), red blood cell casts, or white blood cell casts.
f Renal-related treatment failure was prospectively defined as intake of prohibited medications for adjudicated inadequate lupus nephritis control or renal flare management.
In descriptive subgroup analyses of time to renal-related event or death, results were consistent with the overall endpoint regardless of induction therapy (mycophenolate or cyclophosphamide), biopsy class (Class III or IV, Class III + V or Class IV + V, or Class V; post-hoc analysis), and baseline proteinuria (<3 g/g or ≥3 g/g; post-hoc analysis). The treatment difference was primarily driven by the renal worsening and renal-related treatment failure components of the endpoint.
14.3 Intravenous Administration in Pediatric Patients with SLE
Trial 6: SLE – BENLYSTA 10 mg/kg in Pediatric Patients – Intravenous
The safety and efficacy of BENLYSTA was evaluated in an international, randomized, double-blind, placebo‑controlled, 52-week, pharmacokinetics (PK), efficacy and safety study conducted in 93 pediatric patients with a clinical diagnosis of SLE according to the American College of Rheumatology classification criteria. Patients had active SLE disease, defined as a SELENA-SLEDAI score ≥6 and positive autoantibodies at screening as defined in the adult trials. Patients were on a stable SLE treatment regimen (standard of care) and had similar inclusion and exclusion criteria as in the adult studies. The median age was 15 years (range: 6 to 17). The majority (95%) of patients were female. More than 50% of patients had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA-SLEDAI were mucocutaneous (91%), immunologic (74%), and musculoskeletal (73%). Overall, 19% of pediatric patients had some degree of renal activity and less than 7% had activity in the cardio-respiratory, hematologic, CNS or vascular systems. Randomization into age-related treatment cohorts was stratified by screening SELENA-SLEDAI scores (6 to 12 vs >13) and age (5 to 11 years vs 12 to 17 years).
The primary efficacy endpoint was the SLE Responder Index (SRI-4) at Week 52, as described in the adult intravenous trials. There was a numerically higher proportion of pediatric patients achieving a response in SRI‑4 and its components in pediatric patients receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (Table 8).
Table 8. Pediatric Response Rate at Week 52a (Trial 6):
| Responseb | Placebo + Standard Therapy (n=39) | BENLYSTA 10 mg/kg + Standard Therapy (n=53) |
|---|---|---|
| SLE Responder Index | 44% | 53% |
| Odds Ratio(95% CI) vs. Placebo | 1.49 (0.64, 3.46) | |
| Components of SLE Responder Index | ||
| Percent of patients with reduction in SELENA‑SLEDAI ≥4 | 44% | 55% |
| Percent of patients with no worsening by BILAG index | 62% | 74% |
| Percent of patients with no worsening by PGA | 67% | 76% |
| Other endpoints | ||
| SRI-6 using SELENA SLEDAI ≥6-point reduction | 34% | 41% |
| Proportion of patients with a sustained SRI response | 41% | 43% |
a Based on a non-powered trial.
b Analyses excluded any subject missing a baseline assessment for any of the components (1 for placebo).
Effect on Concomitant Steroid Treatment: At baseline, 95% of pediatric patients were receiving prednisone. Among those pediatric patients, 20% of pediatric patients receiving BENLYSTA plus standard therapy reduced their average prednisone dose by at least 25% per day during Weeks 44 through 52 compared with 21% of pediatric patients on placebo plus standard therapy.
Effect on Severe SLE Flares: In Trial 6, the probability of experiencing a severe SLE flare, as measured by the modified SELENA-SLEDAI Flare Index, excluding severe flares triggered only by an increase of the SELENA-SLEDAI score to >12, was calculated. The proportion of pediatric patients reporting at least one severe flare during the study was numerically lower in pediatric patients receiving BENLYSTA plus standard therapy (17%) compared with those receiving placebo plus standard therapy (35%). Pediatric patients receiving BENLYSTA 10 mg/kg plus standard therapy had a 64% lower risk of experiencing a severe flare during the 52 weeks of observation, relative to the placebo plus standard therapy group. Of the pediatric patients experiencing a severe flare, the median time to the first severe flare was 150 days in pediatric patients receiving BENLYSTA plus standard therapy compared with 113 days in pediatric patients receiving placebo plus standard therapy.
14.4 Subcutaneous Administration in Adults with SLE
Trial 7: SLE – BENLYSTA 200 mg – Subcutaneous
The safety and effectiveness of BENLYSTA administered subcutaneously were evaluated in a randomized, double‑blind, placebo‑controlled trial involving 836 adult patients with SLE according to the American College of Rheumatology criteria (Trial 7, NCT01484496). Patients with severe active lupus nephritis and severe active CNS lupus were excluded. The trial (2:1 randomization) evaluated BENLYSTA 200 mg once weekly plus standard therapy (n=556) compared with placebo once weekly plus standard therapy (n=280) over 52 weeks in patients with active SLE disease. Patients had to have a SELENA-SLEDAI score of ≥8 and positive autoantibody test (anti-nuclear antibody [ANA] and/or anti–double-stranded DNA [anti-dsDNA]) results at screening.
No significant differences in baseline patient characteristics were observed between treatment groups. In some countries, treatment with a B-cell-targeted agent was permitted if received a year or more prior to baseline; otherwise, treatment with a B-cell-targeted agent was not permitted. Patients were excluded from the trial if they were currently receiving other biologic agents. Anti-tumor necrosis factor therapy, intravenous cyclophosphamide, interleukin-1 receptor antagonist, intravenous immunoglobulin (IVIG), prednisone >100 mg/day, and plasmapheresis were not permitted within the previous 3 months or during the trial. The trial was conducted in North America, South America, Europe, and Asia. Baseline concomitant medications included corticosteroids (86%), antimalarials (69%), and immunosuppressives (46%, including azathioprine, methotrexate, and mycophenolate). Most patients (approximately 80%) were receiving 2 or more classes of SLE medications.
More than 50% of patients had 3 or more active organ systems involved at baseline. The most common active organ systems at baseline based on SELENA-SLEDAI were mucocutaneous (88%), musculoskeletal (78%), and immunologic (76%). Overall, 12% of patients had some degree of renal activity and less than 15% of patients had activity in the vascular, cardio-respiratory, or CNS systems. Patients were stratified by disease severity based on their SELENA-SLEDAI score (≤9 vs. ≥10), complement level (C3 and/or C4 low vs. other), and race (Black vs. other), and then randomly assigned to receive BENLYSTA 200 mg plus standard therapy or placebo once weekly plus standard therapy.
The primary efficacy endpoint was the SLE Responder Index-4 (SRI-4) at Week 52 as described in the intravenous trials. Secondary efficacy endpoints included time to first severe flare (as measured by the modified SELENA-SLEDAI SLE Flare Index) and the proportion of patients receiving prednisone >7.5 mg/day at baseline whose average prednisone dose had been reduced by ≥25% to ≤7.5 mg/day during Weeks 40 through 52.
The proportion of patients achieving an SRI-4 response was significantly higher in patients receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy. The trends comparing the treatment groups with respect to the probability of response for the individual components of the endpoint were consistent with that of the SRI-4 (Table 9).
Table 9. Clinical Response Rate in Patients with SLE after 52 Weeks of Treatment (Trial 7):
| Responsea | Placebo + Standard Therapy (n=279) | BENLYSTA + Standard Therapy (n=554) |
|---|---|---|
| SLE Responder Index-4 (SRI-4)b | 48% | 61% P = 0.0006 |
| Odds Ratio (95% CI) vs. placebo | 1.7 (1.3, 2.3) | |
| Components of SLE Responder Index-4 (SRI-4) | ||
| Percent of patients with reduction in SELENA-SLEDAI ≥4 | 49% | 62% |
| Percent of patients with no worsening by BILAG index | 74% | 81% |
| Percent of patients with no worsening by PGA | 73% | 81% |
a Analyses excluded any subject missing a baseline assessment for any of the components (1 for placebo; 2 for belimumab).
b Patients dropping out of the trial early or experiencing certain increases in background medication were considered as failures in these analyses. A higher proportion of patients receiving placebo plus standard therapy were considered as failures for this reason compared with the group receiving BENLYSTA plus standard therapy.
The reduction in disease activity seen in the SRI-4 was related primarily to improvement in the most commonly involved organ systems, namely, mucocutaneous, musculoskeletal, immunologic, and vascular.
The proportion of SRI-4 responders by visit through Week 52 is shown in Figure 3.
Figure 3. Proportion (%) of SRI-4 Responders (+/- Standard Error) by Visita:
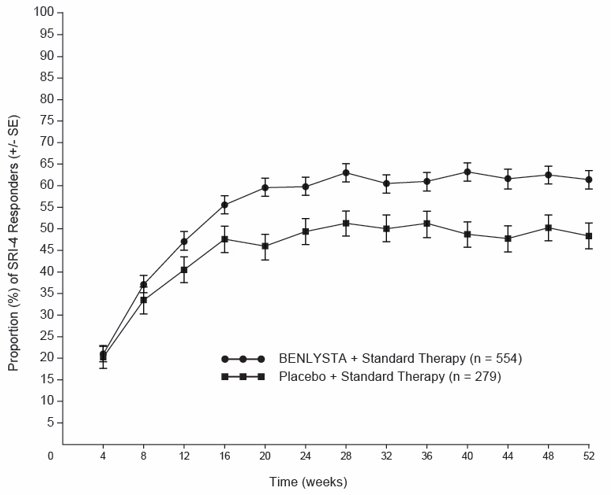
a The same patients may not have responded at each timepoint.
Effect in Black/African-American Patients: Exploratory sub-group analyses of SRI-4 response rate in Black patients (n=91) were performed. The SRI-4 response rate in Black patients receiving BENLYSTA plus standard therapy was 45% (26/58) compared with 39% (13/33) in the group receiving placebo plus standard therapy [see Use in Specific Populations (8.8)].
Effect on Concomitant Steroid Treatment: At baseline, 60% of patients were receiving prednisone at doses >7.5 mg/day. Among these patients, 18% of patients receiving BENLYSTA plus standard therapy reduced their average prednisone dose by at least 25% to ≤7.5 mg/day during Weeks 40 through 52 compared with 12% of patients on placebo plus standard therapy; this difference was not statistically significant (OR = 1.65 [95% CI: 0.95, 2.84]).
Effect on Severe SLE Flares: The probability of experiencing a severe SLE flare, as measured by the modified SELENA-SLEDAI SLE Flare Index, excluding severe flares triggered only by an increase of the SELENA-SLEDAI score to >12, was calculated. The proportion of patients reporting at least 1 severe flare during the study was lower in patients treated with BENLYSTA plus standard therapy (11%) compared with those receiving placebo plus standard therapy (18%). Patients treated with BENLYSTA plus standard therapy had a 49% lower risk of experiencing at least 1 severe flare during the 52 weeks of observation, relative to the patients receiving placebo plus standard therapy (HR = 0.51 [95% CI: 0.35, 0.74]). Of the patients experiencing a severe flare, the median time to the first severe flare was delayed in patients receiving BENLYSTA plus standard therapy compared with placebo plus standard therapy (171 days vs. 118 days).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.