BRAFTOVI Hard capsule Ref.[8687] Active ingredients: Encorafenib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: PIERRE FABRE MEDICAMENT, Les Cauquillous, 81500 Lavaur, France
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein kinase inhibitors
ATC code: L01EC03
Mechanism of action
Encorafenib is a potent and highly selective ATP-competitive small molecule RAF kinase inhibitor. The half maximal inhibitory concentration (IC50) of encorafenib against BRAF V600E, BRAF and CRAF enzymes was determined to be 0.35, 0.47 and 0.30 nM, respectively. The encorafenib dissociation half-life was >30 hours and resulted in prolonged pERK inhibition. Encorafenib suppresses the RAF/MEK/ERK pathway in tumour cells expressing several mutated forms of BRAF kinase (V600E, D and K). Specifically, encorafenib inhibits in vitro and in vivo BRAF V600E, D and K mutant melanoma cell growth and BRAF V600E mutant colorectal cancer cell growth. Encorafenib does not inhibit RAF/MEK/ERK signalling in cells expressing wild-type BRAF.
Combination with binimetinib
Encorafenib and binimetinib (a MEK inhibitor, see section 5.1 of binimetinib SmPC) both inhibit the MAPK pathway, resulting in higher anti-tumour activity, compared to treatment with either drug alone.
Combination with cetuximab
One of the main mechanisms of resistance of BRAF-mutant CRC to RAF inhibitors has been identified as the re-activation of EGFR with bypassing signal transduction via BRAF. Combinations of a BRAF inhibitor, e.g. encorafenib and agents targeting EGFR, e.g. cetuximab have shown to improve anti-tumour efficacy in non-clinical models.
Clinical efficacy and safety
BRAF V600 Mutant Unresectable or Metastatic Melanoma
The safety and efficacy of encorafenib in combination with binimetinib were evaluated in a 2-part Phase III, randomised (1:1:1) active-controlled, open-label, multicentre study in patients with unresectable or metastatic BRAF V600 E or K mutant melanoma (Study CMEK162B2301), as detected using a BRAF assay. Patients had histologically confirmed cutaneous or unknown primary melanoma but those with uveal or mucosal melanoma were excluded. Patients were permitted to receive prior adjuvant therapy and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior treatment with BRAF/MEK inhibitors was not allowed.
Study CMEK162B2301, part 1
In Part 1, patients in the study were randomised to receive encorafenib 450 mg orally daily and binimetinib 45 mg orally twice daily (Combo 450, n=192), encorafenib 300 mg orally daily (Enco 300, n=194), or vemurafenib 960 mg orally twice daily (hereafter referred to as Vem, n=191). Treatment continued until disease progression or unacceptable toxicity. Randomisation was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, vs IVM1c) and Eastern Cooperative Oncology Group (ECOG) performance status (0 vs 1) and prior immunotherapy for unresectable or metastatic disease (yes vs no).
The primary efficacy outcome measure was progression-free survival (PFS) of Combo 450 compared with vemurafenib as assessed by a blinded independent review committee (BIRC). PFS as assessed by investigators (investigator assessment) was a supportive analysis. An additional secondary endpoint included PFS of Combo 450 compared with Enco 300. Other secondary efficacy comparisons between Combo 450 and either vemurafenib or Enco 300 included overall survival (OS), objective response rate (ORR), duration of response (DoR) and disease control rate (DCR) as assessed by BIRC and by investigator assessment.
The median age of patients was 56 years (range 20-89), 58% were male, 90% were Caucasian, and 72% of patients had baseline ECOG performance status of 0. Most patients had metastatic disease (95%) and were Stage IVM1c (64%); 27% of patients had elevated baseline serum lactate dehydrogenase (LDH), and 45% of patients had at least 3 organs with tumour involvement at baseline and 3.5% had brain metastases. 27 patients (5%) had received prior checkpoint inhibitors (anti-PD1/PDL1 or ipilimumab) (8 patients in Combo 450 arm (4%); 7 patients in vemurafenib arm (4%); 12 patients in Enco 300 arm (6%) including 22 patients in the metastatic setting (6 patients in Combo 450 arm; 5 patients in vemurafenib arm; 11 patients in Enco 300 arm) and 5 patients in the adjuvant setting (2 patients in Combo 450 arm; 2 patients in vemurafenib arm; 1 patient in Enco 300 arm).
The median duration of exposure was 11.7 months in patients treated with Combo 450, 7.1 months in patients treated with Enco 300 and 6.2 months in patients treated with vemurafenib. The median relative dose intensity (RDI) for Combo 450 was 100% for encorafenib and 99.6% for binimetinib; the median RDI was 86.2% for Enco 300 and 94.5% for vemurafenib.
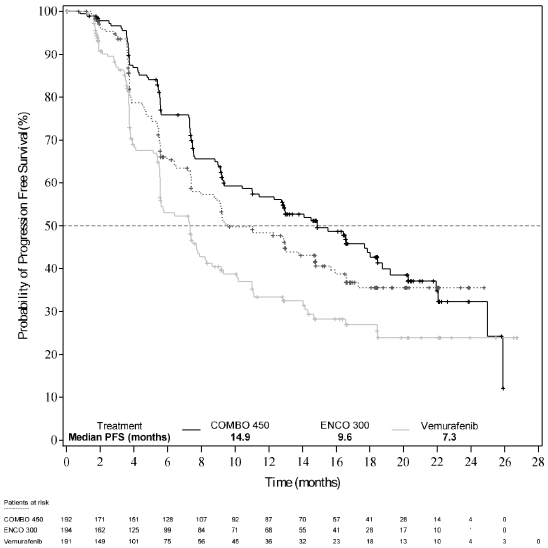
Part 1 of Study CMEK162B2301 demonstrated a statistically significant improvement in PFS in the patients treated with Combo 450 compared with patients treated with vemurafenib. Table 6 and Figure 1 summarise the PFS and other efficacy results based on central review of the data by a blinded independent radiology committee.
The efficacy results based on investigator assessment were consistent with the independent central assessment. Unstratified subgroup analyses demonstrated point estimates in favour of Combo 450, including LDH at baseline, ECOG performance status and AJCC stage.
Table 6. Study CMEK162B2301, Part 1: Progression-free survival and confirmed overall response results (independent central review):
| Encorafenib + binimetinib N=192 (Combo 450) | Encorafenib N=194 (Enco300) | Vemurafenib N=191 (Vem) | |
|---|---|---|---|
| Cut-off date: 19 May 2016 | |||
| PFS (primary analysis) | |||
| Number of events (progressive disease (PD)) (%) | 98 (51.0) | 96 (49.5) | 106 (55.5) |
| Median, months (95% CI) | 14.9 (11.0, 18.5) | 9.6 (7.5,14.8) | 7.3 (5.6, 8.2) |
| HRa (95% CI) (vs Vem) p-value (stratified log-rank)b | 0.54 (0.41, 0.71) <0.001 | ||
| HRa (95% CI) (vs Vem) Nominal p-value | 0.68 (0.52, 0.90) 0.007 | ||
| HRa (95% CI) (vs Enco 300) p-value (stratified log-rank)b | 0.75 (0.56,1.00) 0.051 | ||
| Confirmed overall responses | |||
| Overall response rate, n (%) (95% CI) | 121 (63.0) (55.8, 69.9) | 98 (50.5) (43.3, 57.8) | 77 (40.3) (33.3, 47.6) |
| CR, n (%) | 15 (7.8) | 10 (5.2) | 11 (5.8) |
| PR, n (%) | 106 (55.2) | 88 (45.4) | 66 (34.6) |
| SD, n (%) | 46 (24.0) | 53 (27.3) | 73 (38.2) |
| DCR, n (%) (95% CI) | 177 (92.2) (87.4, 95.6) | 163 (84.0) (78.1, 88.9) | 156 (81.7) (75.4, 86.9) |
| Duration of response | |||
| Median, months (95% CI) | 16.6 (12.2, 20.4) | 14.9 (11.1, NE) | 12.3 (6.9, 16.9) |
| Updated analysis, cut-off date: 07 November 2017 | |||
| PFS | |||
| Number of events (progressive disease) (%) | 113 (58.9) | 112 (57.7) | 118 (61.8) |
| Median, months (95% CI) | 14.9 (11.0, 20.2) | 9.6 (7.4,14.8) | 7.3 (5.6, 7.9) |
| HRa (95% CI) (vs Vem) Nominal p-value | 0.51 (0.39, 0.67) <0.001 | ||
| HRa (95% CI) (vs Vem) Nominal p-value | 0.68 (0.52, 0.88) 0.0038 | ||
| HRa (95% CI) (vs Enco 300) Nominal p-value | 0.77 (0.59,1.00) 0.0498 | ||
CI = Confidence interval; CR = Complete Response; DCR = Disease Control Rate (CR+PR+SD+Non-CR/Non-PD; Non-CR/Non-PD applies only to patients without a target lesion who did not achieve CR or have PD); HR = hazard ratio; NE = Not estimable; PFS = progression-free survival; PR = Partial response; SD = stable disease. Vem = vemurafenib.
a Hazard ratio based on a stratified Cox proportional hazard model
b Log-rank p-value (2-sided)
Figure 1. Study CMEK162B2301, Part 1: Kaplan-Meier plot of progression-free survival by independent central review (cut-off date: 19 May 2016):
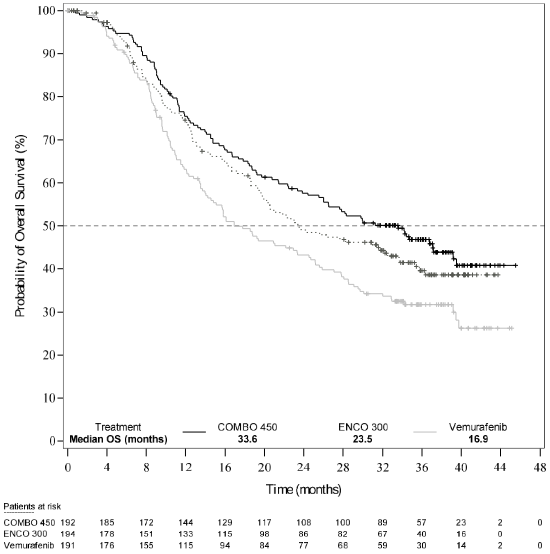
An interim OS analysis of Study CMEK162B2301 Part 1, (cut-off date 07 November 2017) demonstrated a statistically significant improvement in OS for Combo 450 compared with vemurafenib (see Table 7 and Figure 2).
A similar proportion of patients in each treatment arm received subsequent treatment with checkpoint inhibitors, mainly pembrolizumab, nivolumab and ipilimumab (34.4% Combo 450 arm, 36.1% encorafenib arm, 39.8% vemurafenib arm).
Table 7. Study CMEK162B2301, Part 1: Overall survival interim results (cut-off date: 7 November 2017):
| Encorafenib + binimetinib N=192 (Combo 450) | Encorafenib N=194 (Enco 300) | Vemurafenib N=191 (Vem) | |
|---|---|---|---|
| OS | |||
| Number of events (%) | 105 (54.7) | 106 (54.6) | 127 (66.5) |
| Median, months (95% CI) | 33.6 (24.4, 39.2) | 23.5 (19.6, 33.6) | 16.9 (14.0, 24.5) |
| Survival at 12 months (95% CI) | 75.5% (68.8, 81.0) | 74.6% (67.6, 80.3) | 63.1% (55.7, 69.6) |
| Survival at 24 months (95% CI) | 57.6% (50.3, 64.3) | 49.1% (41.5, 56.2) | 43.2% (35.9, 50.2) |
| HR (95% CI) (vs Vem) p-value (stratified log-rank) | 0.61 (0.47, 0.79) <0.0001 | ||
| HR (95% CI) (vs Enco 300) p-value (stratified log-rank) | 0.81 (0.61,1.06) 0.061 | ||
Figure 2. Study CMEK162B2301, Part 1: Kaplan-Meier plot of interim overall survival (cut-off date: 7 November 2017):
Quality of Life (QoL) (cut-off date: 19 May 2016)
The Functional Assessment of Cancer Therapy-Melanoma (FACT-M), the European Organisation for Research and Treatment of Cancer’s core quality of life questionnaire (EORTC QLQ-C30) and the EuroQoL-5 Dimension-5 Level examination (EQ-5D-5L) were used to explore patient-reported outcomes (PRO) measures of health-related Quality of Life, functioning, melanoma symptoms, and treatment-related adverse reactions. A definitive 10% deterioration in FACT-M and in EORTC QLQ-C30 was significantly delayed in patients treated with Combo 450 relative to other treatments. The median time to definitive 10% deterioration in the FACT-M score was not reached in the Combo 450 arm and was 22.1 months (95% CI: 15.2, NE) in the vemurafenib arm with a HR for the difference of 0.46 (95% CI: 0.29, 0.72). An analysis of time to definitive 10% deterioration in EORTC QLQ-C30 score provided with similar results.
Patients receiving Combo 450 reported no change or a slight improvement in the mean change from baseline EQ-5D-5L index score at all visits, whilst patients receiving vemurafenib or encorafenib reported decreases at all visits (with statistical significant differences). An evaluation of change over time in score yielded the same trend for EORTC QLQ-C30 and at all visit for FACT-M.
Study CMEK162B2301, part 2
Part 2 of Study CMEK162B2301 was designed to assess the contribution of binimetinib to the encorafenib and binimetinib combination.
The PFS for encorafenib 300 mg orally daily used in combination with binimetinib 45 mg orally twice daily (Combo 300, n=258) was compared to the PFS for Enco 300 (n=280, including 194 patients from Part 1 and 86 patients from Part 2). Enrolment in Part 2 started after all Part 1 patients were randomised.
Preliminary Part 2 data, at a cut-off date of 9 November 2016, demonstrated the contribution of binimetinib with an improved median PFS estimate of 12.9 months (95% CI: 10.1, 14.0) for Combo 300 compared to 9.2 months (95% CI: 7.4, 11.0) for Enco 300 (Parts 1 and 2) per independent central review (BIRC). Similar results were observed per Investigator assessment. The confirmed ORR per BIRC was 65.9% (95% CI: 59.8, 71.7) for Combo 300 and 50.4% (95% CI: 44.3, 56.4) for Enco 300 (Parts 1 and 2). Median DOR for confirmed responses per BIRC was 12.7 months [95% CI: 9.3, 15.1] for Combo 300 and 12.9 months [95% CI: 8.9, 15.5] for Enco 300. The median duration of treatment was longer for Combo 300 vs Enco 300, 52.1 weeks vs 31.5 weeks.
BRAF V600E Mutant metastatic colorectal cancer – Study ARRAY-818-302
Encorafenib in combination with cetuximab was evaluated in a randomised, active-controlled, open- label, multicentre trial (ARRAY 818-302 BEACON CRC). Eligible patients were required to have BRAF V600E mutant metastatic colorectal cancer that had progressed after 1 or 2 prior regimens. Enrolled patients were eligible to receive cetuximab per locally approved label with regards to tumour RAS status. Prior use of RAF inhibitors, MEK inhibitors or EGFR inhibitors was prohibited. Randomisation was stratified by Eastern Cooperative Oncology Group (ECOG) performance status, prior use of irinotecan and cetuximab source.
A total of 665 patients were randomised (1:1:1) to receive encorafenib 300 mg orally daily in combination with cetuximab dosed as per its approved SmPC (n=220), or encorafenib 300 mg orally daily in combination with binimetinib 45 mg orally twice daily and cetuximab dosed as per its approved SmPC (n=224) or Control (irinotecan with cetuximab or irinotecan/5-fluorouracil/folinic acid (FOLFIRI) with cetuximab, n= 221). Treatment continued until disease progression or unacceptable toxicity.
The efficacy outcome measures were overall survival (OS) and overall response rate (ORR) as assessed by a blinded independent central review committee (BIRC), comparing encorafenib 300 mg in combination with cetuximab versus Control. Other efficacy measures are summarised in Table 8 below.
The median age of patients was 61 years (range 26-91), 47% were male and 83% were white. 51% of patients had baseline ECOG performance status of 0, and 51% received prior irinotecan. 46.8% of patients had at least 3 organs with tumour involvement at baseline.
The median duration of exposure was 3.2 months in patients treated with encorafenib 300 mg in combination with cetuximab, and 1.4 months in patients treated with irinotecan/cetuximab or FOLFIRI/cetuximab (Control arm). In patients treated with the combination of encorafenib 300 mg and cetuximab, the median relative dose intensity (RDI) was 98% for encorafenib and 93.5% for cetuximab. In the control arm, the median RDI was 85.4% for cetuximab, 75.7% for irinotecan and in the subset of patients who received Folinic acid and 5-FU, the median RDI was 75.2% and 75% respectively.
Encorafenib 300 mg in combination with cetuximab demonstrated a statistically significant improvement in OS, ORR and PFS compared to Control. Efficacy results are summarised in Table 8 and Figures 3 and 4.
The efficacy results based on investigator assessment were consistent with the independent central assessment.
Table 8. Study ARRAY-818-302: Efficacy Results:
| Encorafenib with cetuximab | Irinotecan with cetuximab or FOLFIRI with cetuximab (Control) | |
|---|---|---|
| Cut-off date: 11 February 2019 (Primary analysis) | ||
| OS | ||
| Number of patientsa | 220 | 221 |
| Number of events (%) | 93 (42.3) | 114 (51.6) |
| Median, months (95% CI) | 8.4 (7.5-11.0) | 5.4 (4.8, 6.6) |
| HR (95% CI)b,c (vs Control) p-valueb,c | 0.60 (0.41-0.88) 0.0002 | |
| Median duration of follow-up, months (95% CI) | 7.6 (6.4, 9.20) | 7.2 (6.1, 8.1) |
| ORR (per BIRC) | ||
| Number of patientse | 113 | 107 |
| ORR n (%) (95% CI)f | 23 (20.4) (13.4, 29.0) | 2 (1.9) (0.2, 6.6) |
| P-valueb,d,g | <0.0001 | |
| CR, n (%) | 6 (5.3) | 0 |
| PR, n (%) | 17 (15.0) | 2 (1.9) |
| SD, n (%) | 57 (50.4) | 26 (24.3) |
| DCR, n (%) (95% CI)f | 84 (74.3) (65.3, 82.1) | 33 (30.8) (22.3, 40.5) |
| PFS (per BIRC) | ||
| Number of patientsa | 220 | 221 |
| Number of events (%) | 133 (60.5) | 128 (57.9) |
| Median PFS, months (95% CI) | 4.2 (3.7, 5.4) | 1.5 (1.5, 1.7) |
| HR (95% CI)b,c P-valueb,d | 0.40 (0.30, 0.55) <0.0001 | |
| Updated analysis, cut-off date: 15 August 2019 | ||
| OS | ||
| Number of patientsa | 220 | 221 |
| Number of events (%) | 128 (58.2) | 157 (71.0) |
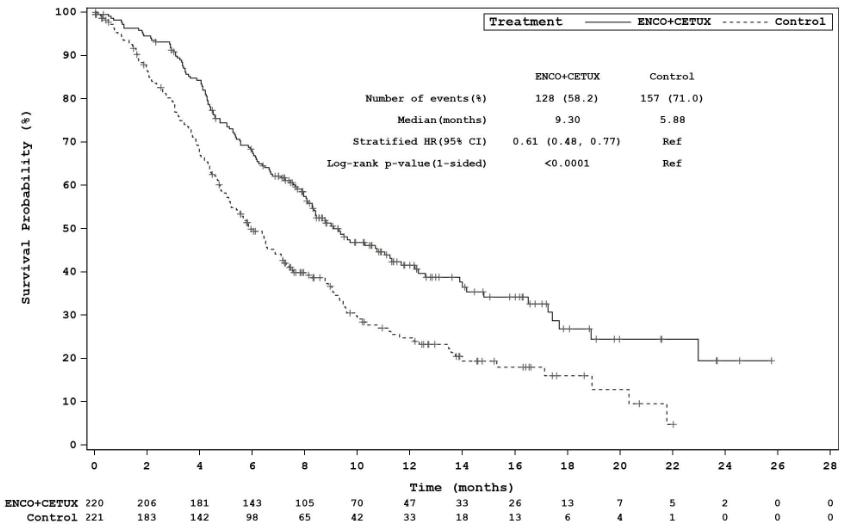
| Median, months (95% CI) | 9.3 (8.0, 11.3) | 5.9 (5.1, 7.1) |
| HR (95% CI)b (vs Control) p-valueb,d,h | 0.61 (0.48, 0.77) <0.0001 | |
| Median duration of follow-up, months (95% CI) | 12.3 (11.1, 14.1) | 12.9 (10.9, 14.6) |
| ORR (per BIRC) | ||
| Number of patientsa | 220 | 221 |
| ORR n (%) (95% CI)f | 43 (19.5) (14.5, 25.4) | 4 (1.8) (0.5, 4.6) |
| p-valueb,d,g,h | <0.0001 | |
| CR, n (%) | 7 (3.2) | 0 |
| PR, n (%) | 36 (16.4) | 4 (1.8) |
| SD, n (%) | 117 (53.2) | 59 (26.7) |
| DCR, n (%) (95% CI)f | 167 (75.9) (69.7, 81.4) | 69 (31.2) (25.2, 37.8) |
| PFS (per BIRC) | ||
| Number of patientsa | 220 | 221 |
| Number of events (%) | 167 (75.9) | 147 (66.5) |
| Median PFS, months (95% CI) | 4.3 (4.1, 5.5) | 1.5 (1.5, 1.9) |
| HR (95% CI)b P-valueb,d,h | 0.44 (0.35, 0.55) <0.0001 | |
CI = Confidence interval; CR = Complete response; HR = Hazard ratio; ORR = Overall response rate; OS = Overall survival; PR = Partial response; SD = Stable disease, DCR: Disease control rate (CR+PR+SD+Non-CR/Non-PD; Non-CR/Non-PD applies only to patients with a non-measurable disease who did not achieve CR or have PD)
a Randomised Phase 3, Full Analysis Set
b Stratified by ECOG PS, source of cetuximab, and prior irinotecan use at randomization
c Repeated CI derived using Lan DeMets O’Brien-Fleming boundaries associated with the observed
information fraction at the interim analysis
d 1-sided
e Among the first 331 randomised patients
f Clopper-Pearson’s method
g Cochran Mantel-Haenszel test
h Nominal p-value
Figure 3. Study ARRAY-818-302: Kaplan-Meier plot of Overall Survival (cut-off date: 11 February 2019):
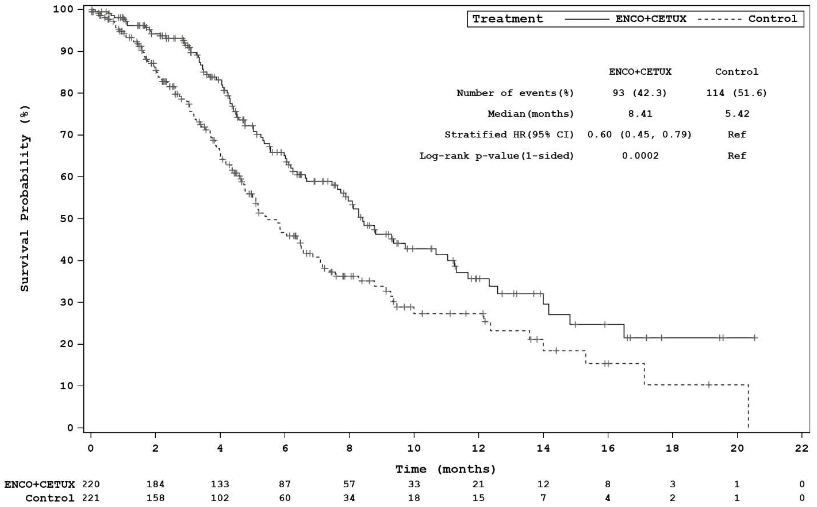
Figure 4. Study ARRAY-818-302: Kaplan-Meier plot of Overall Survival (cut-off date: 15 August 2019):
BRAF V600E Mutant advanced Non-small cell lung cancer – Study ARRAY-818-202
The safety and efficacy of encorafenib in combination with binimetinib were studied in a Phase II, open-label, multicentre, non-comparative study (Study ARRAY-818-202, PHAROS). Patients were required to have histologically-confirmed metastatic NSCLC with a BRAF V600E mutation, ECOG performance status of 0 or 1, and measurable disease. Patients had received 0 or 1 prior line of systemic therapy in the metastatic setting. Prior use of BRAF inhibitors or MEK inhibitors was prohibited.
Patients were enrolled based on the determination of a BRAF V600E mutation in tumour tissue or blood (e.g., ctDNA genetic testing) by a local laboratory assay. Central confirmation of the BRAF V600E mutation status (i.e. any short variant with protein effect V600E) was performed on archival or fresh tumour tissue collected at enrolment and utilized the FoundationOne CDx – F1CDx (tissue) assay.
The analytical sensitivity was assessed through the Limit of Detection (LoD) study for F1CDx using the hit rate method (defined as the lowest level with ≥95% detection) by evaluating variant allele frequency (VAF) for short variants. For F1CDx, the median LoD for substitution was determined to be 3.2% VAF.
A total of 98 patients were enrolled and treated with encorafenib 450 mg orally once daily and binimetinib 45 mg orally twice daily. Treatment continued until disease progression or unacceptable toxicity.
The primary efficacy outcome measure was objective response rate (ORR) and was according to RECIST v1.1 as evaluated by an Independent Radiology Review (IRR). Secondary endpoints included duration of response (DoR), disease control rate (DCR), PFS and OS. Results of the primary analysis with 18.2 months for treatment naïve and 12.8 months previously treated patients are presented below.
Of the 98 patients enrolled in this study, 59 (60.2%) patients were treatment naïve. The median age of patients was 70 years (47-86), 53% were female, 88% were white and 30% had never smoked. 74% had a baseline ECOG performance status of 1 (67.8% of participants had a baseline PS 1 in the treatment naïve population and 82.1% in the previously treated population). All patients had metastatic disease of which 8% had brain metastases at baseline and 97% had adenocarcinoma.
At the time of the primary analysis, the median duration of exposure was 15.1 months in treatment naïve patients and 5.4 months in previously treated patients. In the overall population, the median relative dose intensity (RDI) was 99.2% for encorafenib and 95.4% for binimetinib.
At the time of the primary analysis, the primary endpoint of IRR-assessed ORR in the treatment naïve population was 74.6% (95% CI: 61.6, 85.0), including 9 (15.3%) CRs and 35 (59.3%) PRs. The ORR by IRR in the previously treated population was 46.2% (95% CI: 30.1, 62.8), including 4 (10.3%) CRs and 14 (35.9%) PRs.
Results updated with an additional 10-month follow-up (median duration of exposure of 16.3 months in treatment naïve patients and 5.5 months in previously treated patients) are provided in Table 9.
Table 9. Study ARRAY-818-202: Efficacy Results:
| Encorafenib with Binimetinib | ||
|---|---|---|
| Treatment Naïve (N=59) | Previously Treated (N=39) | |
| ORR per IRR | ||
| ORR, % (95% CI) | 75% (62, 85) | 46% (30, 63) |
| CR, % | 15% | 10% |
| PR, % | 59% | 36% |
| DoR per IRR | N=44 | N=18 |
| Median DoR, months (95% CI) | 40.0 (23.1, NE)* | 16.7 (7.4, NE)* |
| % with DoR ≥12 months | 64% | 44% |
* Results from a sensitivity analysis considering new anti-cancer therapy as an event in addition to progression and death are 23.1 months in treatment naïve patients (14.8; NE) and 12.0 months (6.3; NE) in previously treated patients.
N = number of patients; ORR = Objective Response Rate; CI = Confidence Interval; CR = Complete Response; PR = Partial Response; DoR = Duration of Response; IRR= Independent Radiology Review; NE = not estimable
Cardiac electrophysiology
In the safety analysis of pooled studies, the incidence of new QTcF prolongation >500 ms was 1.1% (4/363) in the Combo 450 ISP (n=372), and 2.5% (5/203) in the encorafenib single agent group of patients with melanoma. QTcF prolongation of >60 ms compared to pre-treatment values was observed in 6.0% (22/364) patients in the Combo 450 ISP, and in 3.4% (7/204) in the encorafenib single agent group (see sections 4.2 and 4.4).
In the safety analysis of the Phase 3 (ARRAY-818-302) safety set in colorectal indication, the incidence of new QTcF prolongation >500 ms was 3.2% (7/216) and QTcF prolongation of >60 ms compared to pre-treatment values was observed in 8.8% (19/216) of patients of the encorafenib + cetuximab arm (see sections 4.2 and 4.4).
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with encorafenib in one or more subsets of the paediatric population in melanoma (see section 4.2 for information on paediatric use).
The European Medicines Agency has waived the obligation to submit the results of studies with encorafenib in all subsets of the paediatric population in colorectal carcinoma and lung cancer (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
The pharmacokinetics of encorafenib were studied in healthy subjects and patients with solid tumours. The pharmacokinetics of encorafenib have been shown to be approximatively dose linear after single and multiples doses. After repeat once-daily dosing, steady-state conditions were reached within 15 days. The accumulation ratio of approximately 0.5 is likely due to auto-induction of CYP3A4. The inter-subject variability (CV%) of AUC is ranged from 12.3% to 68.9%.
Absorption
After oral administration, encorafenib is rapidly absorbed with a median Tmax of 1.5 to 2 hours. Following a single oral dose of 100 mg [14C] encorafenib in healthy subjects, at least 86% of the encorafenib dose was absorbed. Administration of a single 100 mg dose of encorafenib with a high-fat, high-calorie meal decreased the Cmax by 36%, while the AUC was unchanged. A drug interaction study in healthy subjects indicated the extent of encorafenib exposure was not altered in the presence of a gastric pH-altering agent (rabeprazole).
Distribution
Encorafenib is moderately (86.1%) bound to human plasma proteins in vitro. Following a single oral dose of 100 mg [14C] encorafenib in healthy subjects, the mean (SD) blood-to-plasma concentration ratio is 0.58 (0.02) and the mean (CV%) apparent volume of distribution (Vz/F) of encorafenib is 226 L (32.7%).
Biotransformation
Following a single oral dose of 100 mg [14C] encorafenib in healthy subjects, metabolism was found to be the major clearance pathway for encorafenib (approximately 88% of the recovered radioactive dose). The predominant biotransformation reaction of encorafenib was N-dealkylation. Other major metabolic pathways involved hydroxylation, carbamate hydrolysis, indirect glucuronidation and glucose conjugate formation.
Elimination
Following a single oral dose of 100 mg [14C] encorafenib in healthy subjects, radioactivity was eliminated equally in both the faeces and urine (mean of 47.2%). In urine, 1.8% of the radioactivity was excreted as encorafenib. The mean (CV%) apparent clearance (CL/F) of encorafenib was 27.9 L/h (9.15%). The median (range) encorafenib terminal half-life (T1/2) was 6.32 h (3.74 to 8.09 h).
Medicinal product interactions
No drug drug interaction was evidenced between encorafenib and cetuximab.
Effect of CYP enzymes on encorafenib
Encorafenib is metabolised by CYP3A4, CYP2C19 and CYP2D6. In vitro, CYP3A4 was predicted to be the major enzyme contributing to total oxidative clearance of encorafenib in human liver microsomes (~83.3%), followed by CYP2C19 and CYP2D6 (~16.0% and 0.71%, respectively). The effect of co-administering a strong CYP3A4 inducer on encorafenib exposure has not been studied in a dedicated trial. Repeat dose administration of encorafenib 450 mg once daily and binimetinib 45 mg twice daily in melanoma patients with modafinil, a moderate CYP3A4 inducer, decreased encorafenib steady-state AUC by 24% and Cmax by 20%, compared to encorafenib alone.
Effect of encorafenib on CYP substrates
In vitro experiments indicate encorafenib is a relatively potent reversible inhibitor of UGT1A1, CYP2B6, CYP2C9 and CYP3A4/5, as well as a time-dependent inhibitor of CYP3A4. Encorafenib induced CYP1A2, CYP2B6, CYP2C9 and CYP3A4 in human primary hepatocytes.
Repeat dose administration of encorafenib 450 mg once daily and binimetinib 45 mg twice daily in melanoma patients with a single dose of CYP probe substrate cocktail reduced midazolam 2 mg (CYP3A4 substrate) AUC by 82% and Cmax by 74%. It decreased omeprazole 20 mg (CYP2C19 substrate) AUC by 17% and did not change Cmax and increased caffeine 50 mg (CYP1A2 substrate) AUC by 27% and Cmax by 13%. It decreased the ratio of losartan metabolite E3174 to losartan (CYP2C9 substrate) concentrations in urine by 28% and did not change the ratio of dextromethorphan metabolite (dextrorphan) to dextromethorphan (CYP2D6 substrate) concentrations in urine. These results indicate strong induction of CYP3A4, mild inhibition of CYP1A2 and no impact on pharmacokinetics of CYP2C19 substrates. From the urinary data, the inhibitory potency on CYP2C9 and CYP2D6 cannot be finally concluded. No data are available for CYP2D6 poor metabolisers.
A single dose of encorafenib 450 mg and binimetinib 45 mg reduced bupropion 75 mg (CYP2B6 substrate) AUC and Cmax by ≤25%. Repeated administration of encorafenib 450 mg daily and binimetinib 45 mg twice daily reduced bupropion AUC and Cmax by 26 % and increased the AUC of the active metabolite hydroxybupropion by 49% indicating mild induction.
For co-administration with UGT1A1 substrates that undergo gut extraction, a minor to moderate interaction is expected. While binimetinib is a UGT1A1 substrate, it does not undergo gut extraction and therefore no DDI with encorafenib is expected. Additionally, no differences in exposure have been observed clinically when binimetinib is co-administered with encorafenib.
Effect of transporters on encorafenib
Encorafenib was found to be a substrate of the P-glycoprotein (P-gp) transporters. Inhibition of P-gp is unlikely to result in a clinically important increase in encorafenib concentrations as encorafenib exhibits high intrinsic permeability. The involvement of several uptake transporter families (OCT1, OATP1B1, OATP1B3 and OATPB1) was investigated in vitro using relevant transporter inhibitors. The data suggest that hepatic uptake transporters are not involved in encorafenib distribution into primary human hepatocytes.
Effect of encorafenib on transporters
Repeated administration of encorafenib 450 mg once daily and binimetinib 45 mg twice daily with a single dose of rosuvastatin (a OATP1B1, OATP1B3 and BCRP substrate) increased rosuvastatin Cmax by 2.7-fold and AUC by 1.6-fold indicating a mild inhibition of OATP1B1, OATP1B3 and/or BCRP transporters.
In vitro, encorafenib inhibited the hepatic transporter OCT1, but is unlikely to be an effective inhibitor clinically. Based on in vitro studies, there is potential for encorafenib to inhibit renal transporters OCT2, OAT1, OAT3 at clinical concentrations. In addition, encorafenib may inhibit P-gp in the gut at the expected clinical concentrations.
Special populations
Age
Based on a population pharmacokinetic analysis, age was found to be a significant covariate on encorafenib volume of distribution, but with high variability. Given the small magnitude of these changes and high variability, these are unlikely to be clinically meaningful, and no dose adjustments are needed for elderly patients.
Gender
Based on a population pharmacokinetic analysis gender was not found to be a significant model covariate on clearance or volume of distribution. As a result, no major changes in encorafenib exposure are expected based upon gender. Body weight Based on a population pharmacokinetic analysis, body weight was found to be a significant model covariate on clearance and volume of distribution. However, given the small magnitude of change in clearance and the high variability in the predicted volume of distribution in the model, weight is unlikely to have a clinically relevant influence on the exposition of encorafenib.
Race
There are no clinically relevant differences in encorafenib PK between Asians and non Asians. There are insufficient data to evaluate potential differences in the exposure of encorafenib in other races or ethnicity.
Hepatic impairment
Results from a dedicated clinical study indicate a 25% higher total encorafenib exposures in patients with mild hepatic impairment (Child-Pugh Class A) compared with subjects with normal liver function. This translates into a 55% increase of the unbound encorafenib exposure.
The pharmacokinetics of encorafenib has not been evaluated clinically in patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment. As encorafenib is primarily metabolised and eliminated via the liver, based on PBPK modelling, patients with moderate to severe hepatic impairment may have greater increases in exposure than patients with mild hepatic 33 impairment. No dosing recommendation can be made in patients with moderate or severe hepatic impairment (see sections 4.2 and 4.4).
Renal impairment
Encorafenib undergoes minimal renal elimination. No formal clinical study has been conducted to evaluate the effect of renal impairment on the pharmacokinetics of encorafenib.
In a population pharmacokinetic analysis, no clear trend in encorafenib CL/F was observed in patients with mild (eGFR 60 to 90 mL/min/1.73 m²) or moderate (eGFR 30 to 59 mL/min/1.73 m²) renal impairment compared with subjects with normal renal function (eGFR ≥90 mL/min/1.73 m²). A small decrease in CL/F (≤5%) was predicted for patients with mild and moderate renal impairment, which is unlikely to be clinically relevant. The pharmacokinetics of encorafenib have not been studied in patients with severe renal impairment.
Preclinical safety data
In the 4-week and 13-week rat toxicity studies, clinical signs, reduced body weight reduced epididymides and prostate weights and microscopic findings in testes, epididymides, stomach and skin were noted. Partial reversibility of these findings was noted after a 4-week recovery period. No NOAEL could be established for the 4-week study. The NOAEL determined in the 13-week study was more than 10-times human therapeutic exposures.
In the 4-week and 13-week monkey toxicity study, isolated/sporadic episodes of emesis and diarrhoea as well as ophthalmic lesions were observed at slightly above human therapeutic exposures. Ophthalmic lesions were partially reversible and consisted of a separation or detachment in the retina between the outer rods and cones layer and retinal pigmented epithelium at the central macula at the fovea. This observation was similar to that described in humans as central serous-like chorioretinopathy or central serous retinopathy.
Encorafenib was not genotoxic.
Fertility studies were not conducted with encorafenib. In the 13-week rat toxicology studies, encorafenib treatment at 6 mg/kg/d (dose level more than 5 times the human exposure at the therapeutic dose) resulted in decreased testes and epididymis weights with tubular degeneration and oligospermia. In the 13-week study, partial reversibility was noted at the highest dose level (60 mg/kg/d).
The embryo-foetal development study in rats indicated that encorafenib induced foetal toxicity with lower foetal weights and delays in skeletal development.
The embryo-foetal development study in rabbits indicated that encorafenib induced foetal toxicity with lower foetal weights and transitory changes in skeletal development. Dilatation of the aortic arc was observed in some foetuses.
Encorafenib was phototoxic in an in vitro 3T3 Neutral Red Uptake Test. Encorafenib was not a sensitiser in the in vivo mouse sensitization assay. Collectively, these data indicate that encorafenib has a risk of phototoxic potential and minimal risk for sensitization at therapeutic doses in patients.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.