BRENZAVVY Tablet Ref.[107226] Active ingredients: Bexagliflozin
Source: FDA, National Drug Code (US) Revision Year: 2023
12.1. Mechanism of Action
Bexagliflozin is an inhibitor of sodium-glucose co-transporter 2 (SGLT2), the transporter responsible for reabsorption of the majority of glucose from the renal glomerular filtrate in the renal proximal tubule. By inhibiting SGLT2, bexagliflozin reduces renal reabsorption of filtered glucose and lowers the renal threshold for glucose, and thereby increases urinary glucose excretion.
12.2. Pharmacodynamics
Urinary Glucose Excretion and Urinary Volume
Dose-dependent increases in urinary glucose excretion (UGE) accompanied by increases in urine volume were observed in healthy subjects and in adults with type 2 diabetes mellitus following single- and multiple-dose administration of bexagliflozin. Dose-response analysis indicates that 20 mg bexagliflozin provides near-maximal UGE. Elevated UGE was maintained after multiple-dose administration.
Cardiac Electrophysiology
At 5 times the recommended dose, bexagliflozin does not prolong the QTc interval to any clinically significant extent.
12.3. Pharmacokinetics
The pharmacokinetics of bexagliflozin are similar in healthy subjects and adults with type 2 diabetes mellitus. Following dosing in the fasted state, mean Cmax and AUC0-∞ were 134 ng/mL and 1,162 ng·h/mL, respectively. Bexagliflozin does not exhibit time-dependent pharmacokinetics and accumulates in plasma up to ~20% following multiple dosing.
Absorption
Following oral administration of BRENZAVVY, peak plasma concentrations of bexagliflozin were reached between 2–4 hours post-dose and can be delayed if taken after a meal or by medications that slow gastric emptying. Plasma Cmax and AUC of bexagliflozin increase in a dose-proportional manner following single doses from 3 mg (0.15 times the recommended dose) to 90 mg (4.5 times the recommended dose).
Effect of Food
Administration of BRENZAVVY after consumption of a standard high-fat, high-caloric meal increased Cmax and AUC by 31% and 10%, respectively, compared to dosing in the fasted state. The median Tmax was increased to 5 hours. The effects of food on bexagliflozin pharmacokinetics are not considered clinically relevant [see Dosage and Administration (2.2)].
Distribution
Bexagliflozin is approximately 93% bound to plasma protein. Neither renal nor hepatic impairment substantially alters protein binding. The apparent volume of distribution is 262 L.
Elimination
Metabolism
Bexagliflozin is mainly metabolized by UGT1A9 and, to a lesser extent, CYP3A. In plasma the most abundant metabolite is the pharmacologically inactive 3′-O-glucuronide, which was found to constitute 32.2% of the parent compound AUC in a radiolabeled tracer study. None of the metabolites are expected to have clinically relevant pharmacological effects.
Excretion
The apparent oral clearance of bexagliflozin is 19.1 L/h by population pharmacokinetic modeling. The apparent terminal elimination half-life of bexagliflozin was approximately 12 hours. Following administration of an oral [14C]-bexagliflozin solution to healthy subjects, 91.6% of input radioactivity was recovered, 51.1% in feces, the majority as bexagliflozin, and 40.5% in urine, largely as the 3′-O-glucuronide. The proportion of input radioactivity recovered as bexagliflozin in urine and feces was 1.5% and 28.7%, respectively
Specific Populations
Patients with Renal Impairment
In a clinical pharmacology study in patients with type 2 diabetes mellitus and mild (eGFR 60 to 89 mL/min/1.73 m²), moderate (eGFR 30 to 59 mL/min/1.73 m²), and severe (eGFR less than 30 mL/min/1.73 m²) renal impairment, the AUC of bexagliflozin was 7%, 34% and 54% greater than in patients with normal renal function, respectively, after administration of a single 20 mg dose of BRENZAVVY. These increases in bexagliflozin AUC are not considered clinically meaningful.
Consistent with the mechanism of action of bexagliflozin, the 24-hour UGE in patients with type 2 diabetes mellitus and mild, moderate, and severe renal impairment was 17%, 60%, and 83% lower than in patients with type 2 diabetes mellitus with normal renal function, respectively. Therefore, the glucose-lowering pharmacodynamic response to bexagliflozin declines with increasing severity of renal impairment [see Dosage and Administration (2.1), Warnings and Precautions (5.3), Use in Specific Populations (8.6), and Clinical Studies (14)]. The impact of hemodialysis on bexagliflozin exposure is not known.
Patients with Hepatic Impairment
In patients with moderate hepatic impairment (Child-Pugh class B), the AUC of bexagliflozin increased by 28%, and Cmax increased by 6.3% compared to subjects with normal hepatic function. These increases in bexagliflozin AUC and Cmax are not considered clinically meaningful. There is no clinical experience in patients with Child-Pugh class C (severe) hepatic impairment [see Use in Specific Populations (8.7).
Effects of Age, Body Weight, Sex, and Race
Based on a population pharmacokinetic analysis, age, body weight, sex and race do not have a clinically relevant effect on the pharmacokinetics of bexagliflozin.
Drug Interaction Studies
In vitro Assessment of Drug Interactions
Based on in vitro studies, bexagliflozin is not expected to inhibit CYP450 isoenzymes (CYPs) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4, or induce CYPs 1A2, 2C19 and 3A4 at clinically relevant plasma concentrations.
Bexagliflozin is not expected to inhibit drug transporters including breast cancer resistance protein (BRCP), bile salt export pump (BSEP), organic anion transporting polypeptides (OATP1B1, OATP1B3), anion transporters (OAT1, OAT3), organic cation transporters (OCT1, OCT2), and multidrug and toxin extrusion transporters (MATE1, MATE2-K) at clinically relevant plasma concentrations. Bexagliflozin is a substrate for P-glycoprotein (P-gp).
In vivo Assessment of Drug Interactions
There are no clinically meaningful changes in bexagliflozin exposure when taken with metformin, glimepiride, sitagliptin, exenatide, probenecid, or verapamil (Figure 1). Bexagliflozin had no clinically relevant effect on the pharmacokinetics of metformin, glimepiride, sitagliptin, and digoxin (Figure 2).
Figure 1. Effect of Other Drugs on the Pharmacokinetics of Bexagliflozin:
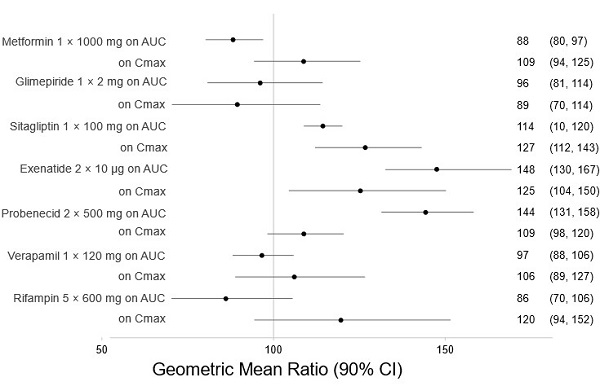
Note: Rifampin dosing over 5 days, the regimen may not represent the maximal impact on bexagliflozin exposure. All others were single day dosing (once daily or twice daily).
Figure 2. Effect of Bexagliflozin on the Pharmacokinetics of Other Drugs

Note: Single day dosing.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenesis was evaluated in 2-year studies in CD-1 mice at oral gavage doses of 15, 50, and 150 mg/kg/day and in Sprague-Dawley (SD) rats at 3, 10, and 30 mg/kg/day. In male rats, the dose was reduced to 1, 3, and 10 mg/kg/day at week 73 due to exacerbation of chronic progressive nephropathy resulting in excessive mortality. There were no drug-related neoplastic findings in mice or rats at up to the highest doses tested representing up to 156 times (mice) and 68 times (rats) the clinical dose of 20 mg based on AUC.
Mutagenesis
Bexagliflozin was not mutagenic or clastogenic with or without metabolic activation in the in vitro Ames bacterial mutagenicity assay, the in vitro CHO cell assay, an in vivo micronucleus assay in rats, and an in vivo unscheduled hepatic DNA synthesis study in rats.
Impairment of Fertility
Bexagliflozin had no effects on mating, fertility or early embryonic development in male or female rats at any dose up to the highest dose of 200 mg/kg/day, which resulted in exposures 280 times (males) and 439 times (females) the 20 mg clinical dose (based on AUC).
14. Clinical Studies
14.1 Overview of Clinical Trials
BRENZAVVY has been studied as monotherapy (Trial 1) and in combination with metformin in adults with type 2 diabetes mellitus (Trials 2, 3, and 4) [see Clinical Studies (14.2 and 14.3)]. BRENZAVVY has also been studied in adults with type 2 diabetes mellitus with moderate renal impairment (Trial 5) [see Clinical Studies (14.4)], and in adults with type 2 diabetes mellitus with established CVD or at increased risk for CVD (Trial 6) [see Clinical Studies (14.5)].
Treatment with BRENZAVVY reduced hemoglobin A1c (HbA1c) compared to placebo and efficacy was noninferior to glimepiride (up-titrated to a maximum dose of 6 mg) and sitagliptin 100 mg once daily (see Trials 3 and 4). The reduction in HbA1c by BRENZAVVY was shown across subgroups of age, sex, race, and geographic region.
14.2 Glycemic Control in Adults with Type 2 Diabetes Mellitus - Monotherapy (Trial 1)
A total of 207 adults with type 2 diabetes mellitus inadequately controlled (HbA1c between 7% and 10.5%) by diet and exercise participated in a randomized, double-blind, multi-center, 24-week, placebo-controlled trial (NCT02715258; referred to as Trial 1) to evaluate the efficacy of BRENZAVVY monotherapy. Patients were either treatment naïve or had discontinued a single oral antihyperglycemic treatment ≥6 weeks prior to entering a 2-week, single-blind, placebo run-in period. Upon completion of the run-in period they were randomized (1:2) to placebo or BRENZAVVY 20 mg administered orally once daily. The mean age of the population was 55 years and 4% of the patients were older than 75 years of age. Forty-eight percent (48%) were male and 74% were White, 10% were Asian, 15% were Black and 1% were other races. Fifty-two percent (52%) were Hispanic/Latino.
At week 24, treatment with BRENZAVVY provided a statistically significant reduction in HbA1c compared to placebo (see Table 5).
Table 5. Glycemic Results from a 24-Week Placebo-Controlled Monotherapy Trial of BRENZAVVY in Adults with Type 2 Diabetes Mellitus (Trial 1):
| Placebo N=69 | BRENZAVVY N=138 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 7.9 | 8.1 |
| Change from baseline [adjusted mean (SE)]a | -0.1 (0.1) | -0.5 (0.1) |
| Difference from placebo [adjusted mean] (95% CI) | -0.4 (-0.6, -0.1) * | |
| Proportion of patients (%) achieving HbA1c <7%b | 20% | 31% |
| FPG (mg/dL) | ||
| Baseline mean | 170 | 169 |
| Change from baseline [adjusted mean (SE)]c | -3 (4) | -16 (3) |
| Difference from placebo [adjusted mean] (95% CI) | -14 (-24, -3) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
* Statistically significant (multiplicity adjusted one-sided p-value <0.025)
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 24 (9% and 7% for bexagliflozin and placebo, respectively). The ANCOVA model included treatment, country, background anti-diabetes treatment status (treatment naïve or not) and the baseline HbA1c value as a covariate.
b Crude proportion using imputed HbA1c values for missing data at week 24 and averaged across multiply imputed datasets
c Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
Figure 3. Mean Change from Baseline in HbA1c (%) by Time and Treatment:
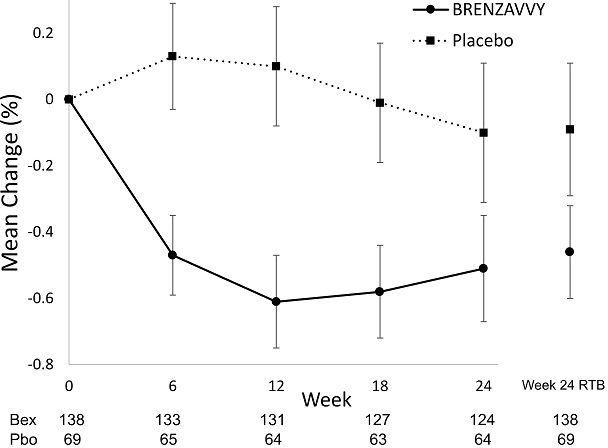
Vertical lines represent 95% confidence intervals. Treatment group least squares mean changes from baseline in HbA1c were estimated by an ANCOVA model using observed data for intermediate visits. Numbers of patients per arm per measurement shown below plot. Return-to-baseline (RTB) analysis results at Week 24 are plotted to the right separately.
14.3 Glycemic Control in Adults with Type 2 Diabetes Mellitus – Combination Therapy (Trials 2, 3 and 4)
Add-on Combination Therapy with Metformin (Trial 2)
A total of 317 adults with type 2 diabetes mellitus inadequately controlled (HbA1c between 7.5% and 10.5%) by metformin monotherapy (≥1,000 mg/day or ≥1,500 mg/day for ≥8 weeks depending on country) participated in a randomized, double-blind, multi-center, 24-week, placebo-controlled trial (NCT03259789; referred to as Trial 2) to evaluate the efficacy of BRENZAVVY in combination with metformin. Patients entered a 1-week, single-blind, placebo run-in period, and were randomized (1:1) to placebo or BRENZAVVY 20 mg administered orally once daily in addition to the background metformin therapy. The mean age of the population was 56 years and 4% of the patients were older than 75 years of age. Sixty-one percent (61%) were male and 31% were White, 50% were Asian, 17% were Black and 2% were other races. Twenty-one percent (21%) were Hispanic/Latino.
At Week 24, treatment with BRENZAVVY provided a statistically significant reduction in HbA1c compared to placebo (see Table 6).
Table 6. Glycemic Results From a 24-Week Placebo-Controlled Trial for BRENZAVVY used in Combination with Metformin in Adults with Type 2 Diabetes Mellitus (Trial 2):
| Placebo N=159 | BRENZAVVY N=158 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 8.5 | 8.6 |
| Change from baseline [adjusted mean (SE)]a | -0.5 (0.1) | -1.0 (0.1) |
| Difference from placebo [adjusted mean] (95% CI) | -0.5 (-0.7, -0.3)* | |
| Proportion of patients (%) achieving HbA1c <7%b | 10% | 26% |
| FPG (mg/dL) | ||
| Baseline mean | 190 | 186 |
| Change from baseline [adjusted mean (SE)]c | -20 (3) | -42 (3) |
| Difference from placebo [adjusted mean] (95% CI) | -22 (-31, -12) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
* Statistically significant (multiplicity adjusted one-sided p-value <0.025)
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 24 (10% and 9% for bexagliflozin and placebo, respectively). The ANCOVA model included treatment, baseline HbA1c value and country (US or Japan).
b Crude proportion using imputed HbA1c values for missing data at week 24 and averaged across multiply imputed datasets
c Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
Active-Controlled Trial versus Glimepiride as Add-on Therapy with Metformin (Trial 3)
A total of 426 adults with type 2 diabetes mellitus inadequately controlled (HbA1c between 7% and 10.5%) by metformin monotherapy participated in a randomized, double-blind, multi-center, 60-week, active comparator-controlled trial (NCT02769481; referred to as Trial 3) to evaluate the efficacy of BRENZAVVY in combination with metformin. Patients receiving metformin (≥1,500 mg/day) as monotherapy entered a 2-week, single-blind, placebo run-in period; patients receiving metformin and another oral hypoglycemic agent enrolled if they discontinued the second agent for at least 6 weeks. Upon completion of the run-in period, they were randomized (1:1) to glimepiride or BRENZAVVY 20 mg administered orally once daily in addition to the background metformin therapy. Glimepiride therapy was initiated at 2 mg/day and titrated up to 6 mg/day or the maximum tolerated dose below 6 mg/day. Glimepiride up-titration occurred until week 6 of the 60-week treatment period. The mean daily dose of glimepiride was 5.4 mg (the maximal approved dosage in the United States is 8 mg per day). Eighty-one percent of patients in the glimepiride group were titrated up to 6 mg per day. The mean age of the population was 60 years and 5% of the patients were older than 75 years of age. Fifty-eight percent (58%) were male and 94% were White, 3% were Asian, 2% were Black, and 1% were other races. Twenty-two percent (22%) were Hispanic/Latino.
Glycemic Results:
BRENZAVVY was non-inferior to glimepiride in the change in HbA1c from baseline after 60 weeks of treatment (See Table 7).
Table 7. Glycemic Results from a 60-Week Active-Controlled Trial Comparing BRENZAVVY to Glimepiride as an Add-On Therapy in Adults with Type 2 Diabetes Mellitus Inadequately Controlled by Metformin (Trial 3):
| Glimepiride N=213 | BRENZAVVY N=213 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 8.0 | 8.0 |
| Change from baseline [adjusted mean (SE)]a | -0.6 (0.1) | -0.7 (0.1) |
| Difference from glimepiride [adjusted mean] (95% CI) | -0.0 (-0.2, 0.1)b | |
| Proportion of patients (%) achieving HbA1c <7%c | 33% | 35% |
| FPG (mg/dL) | ||
| Baseline mean | 174 | 172 |
Change from baseline [adjusted mean (SE)]d | -14 (3) | -22 (2) |
| Difference from glimepiride [adjusted mean] (95% CI) | -8 (-15, -1) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 60 (9% and 10% for bexagliflozin and glimepiride, respectively). The ANCOVA model included treatment, region, background treatment status (metformin-only or metformin + another oral hypoglycemic agent), eGFR at baseline (≥90 vs. <90 mL/min/1.73 m²), and baseline HbA1c value. Non-inferiority is declared if the upper bound of the 95% confidence interval for the difference from glimepiride lies below 0.35.
b Non-inferior
c Crude proportion using imputed HbA1c values for missing data at week 60 and averaged across multiply imputed datasets
d Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
Body Weight and Systolic Blood Pressure Results:
The mean baseline body weight in patients with baseline BMI ≥25 kg/m² was 92 kg (N=202) and 90 kg (N=201) in the glimepiride and BRENZAVVY groups, respectively. The mean changes from baseline to Week 60 in this population were 0.6 kg and -3.4 kg in the glimepiride and BRENZAVVY groups, respectively. The difference from glimepiride (95% CI) for BRENZAVVY was -4.0 kg (-4.8, -3.2).
The mean baseline cuff systolic blood pressure (SBP) in patients with baseline values ≥140 mmHg was 149 mm Hg in the glimepiride (N=75) and BRENZAVVY (N=78) groups. The mean changes in cuff SBP in this population from baseline to Week 60 were -6.2 mmHg and -12.9 mmHg in the glimepiride and BRENZAVVY groups, respectively. The difference from glimepiride (95% CI) for BRENZAVVY was -6.8 mmHg (-10.8, -2.6).
Active-Controlled Trial versus Sitagliptin as Add-on Therapy with Metformin (Trial 4)
A total of 384 adults with type 2 diabetes mellitus inadequately controlled (HbA1c between 7% and 11%) by metformin monotherapy participated in a randomized, double-blind, multi-center, 24-week, active comparator-controlled trial (NCT03115112; referred to as Trial 4) to evaluate the efficacy of BRENZAVVY in combination with metformin. Patients receiving metformin monotherapy (≥1,500 mg/day for ≥8 weeks) entered a 1-week, single-blind, placebo run-in period and were randomized to sitagliptin 100 mg or BRENZAVVY 20 mg administered orally once daily in addition to the background metformin therapy. The mean age of the population was 59 years and 4% of the patients were older than 75 years of age. Sixty-four percent (64%) were male and 82% were White, 16% were Asian, and 2% were Black. Three percent (3%) were Hispanic/Latino.
BRENZAVVY was non-inferior to sitagliptin in the change in HbA1c from baseline after 24 weeks of treatment.
Table 8. Glycemic Results from a 24-Week Active-Controlled Trial Comparing BRENZAVVY to Sitagliptin as an Add-On Therapy in Adults with Type 2 Diabetes Mellitus Inadequately Controlled by Metformin (Trial 4):
| Sitagliptin N=193 | BRENZAVVY N=191 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 8.0 | 7.9 |
| Change from baseline [adjusted mean (SE)]a | -0.9 (0.1) | -0.8 (0.1) |
| Difference from sitagliptin [adjusted mean] (95% CI) | 0.1 (-0.1, 0.2)b | |
| Proportion of patients (%) achieving HbA1c <7%c | 45% | 40% |
| FPG (mg/dL) | ||
| Baseline mean | 180 | 176 |
| Change from baseline [adjusted mean (SE)]d | -26 (2) | -31 (2) |
| Difference from sitagliptin [adjusted mean] (95% CI) | -5 (-11, 1) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 24 (6% and 2% for bexagliflozin and sitagliptin, respectively). The ANCOVA model included treatment, baseline HbA1c value and region. Non-inferiority is declared if the upper bound of the 95% confidence interval for the difference from sitagliptin lies below 0.35.
b Non-inferior
c Crude proportion using imputed HbA1c values for missing data at week 24 and averaged across multiply imputed datasets
d Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
14.4 Glycemic Control in Adults with Type 2 Diabetes Mellitus and Moderate Renal Impairment (Trial 5)
A total of 312 adults with inadequately controlled type 2 diabetes mellitus (HbA1c between 7.0% and 10.5%) and moderate renal impairment (eGFR between 30 and 60 mL/min/1.73 m²) received BRENZAVVY or placebo in a double-blind, randomized, placebo-controlled trial (NCT02836873; referred to as Trial 5) to evaluate the efficacy of BRENZAVVY. In this trial, 77 BRENZAVVY-treated patients had an eGFR between 45 and 60 mL/min/1.73 m² and 74 BRENZAVVY-treated patients had an eGFR between 30 and 45 mL/min/1.73 m². At baseline, nearly all patients (96%) were treated with one or more antidiabetic medications including metformin (37%), insulin (58%), sulfonylureas (11%) and dipeptidyl peptidase 4 (DPP-4) inhibitors (21%). The mean age of the population was 70 years and 29% of the patients were older than 75 years of age. Sixty-three percent (63%) were male and 55% were White, 38% were Asian, 5% were Black, and 2% were other races. Eight percent (8%) were Hispanic/Latino. The mean duration of type 2 diabetes mellitus was 16 years, the mean HbA1c at baseline was 8.0% and the mean eGFR was 45 mL/min/1.73 m².
At week 24, treatment with BRENZAVVY provided a statistically significant reduction in HbA1c compared to placebo (Table 9).
Table 9. Glycemic Results from a 24-Week Placebo-Controlled Trial that Evaluated BRENZAVVY as a Therapy Added to Standard of Care Regimens for Adults with Type 2 Diabetes Mellitus and eGFR between 30 and 60 mL/min/1.73 m² (Trial 5):
| Placebo N=155 | BRENZAVVY N=157 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 7.9 | 8.0 |
| Change from baseline [adjusted mean (SE)]a | -0.3 (0.1) | -0.6 (0.1) |
| Difference from placebo [adjusted mean] (95% CI) | -0.3 (-0.4, -0.1)* | |
| Proportion of patients (%) achieving HbA1c <7%b | 22% | 33% |
| FPG (mg/dL) | ||
| Baseline mean | 155 | 156 |
| Change from baseline [adjusted mean (SE)]c | -8 (3) | -22 (3) |
| Difference from placebo [adjusted mean] (95% CI) | -14 (-23, -5) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
* Statistically significant (multiplicity adjusted one-sided p-value <0.025)
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 24 (3% and 5% for bexagliflozin and placebo, respectively). The ANCOVA model included treatment, region, screening anti-diabetic treatment regimen (insulin treated or other), baseline eGFR (<45 or ≥45 mL/min/1.73m²) and baseline HbA1c value.
b Crude proportion using imputed HbA1c values for missing data at week 24 and averaged across multiply imputed datasets
c Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
Body Weight Results
The mean baseline body weight in patients with baseline BMI ≥25 kg/m² was 89 kg in both the placebo (N=122) and BRENZAVVY (N=125) groups. The mean changes from baseline to Week 24 in this population were -0.4 kg and -2.1 kg in the placebo and BRENZAVVY groups, respectively. The difference from placebo (95% CI) for BRENZAVVY was -1.7 kg (-2.4, -1.0).
14.5 Glycemic Control and Major Cardiovascular Events (MACE) in Adults with Type 2 Diabetes Mellitus and Increased Risk for Cardiovascular Disease (Trial 6)
The efficacy of BRENZAVVY was assessed in a multicenter, randomized, double-blind, placebo-controlled trial (NCT02558296; referred to as Trial 6) of adults with inadequately controlled type 2 diabetes mellitus (HbA1c between 7% and 11%) who had either established CVD (including a history of atherosclerotic vascular disease or a history of heart failure) or multiple risk factors for CVD. There were no restrictions on background antihyperglycemic medication use, aside from treatment with an SGLT2 inhibitor. After a single-blind, 2-week, placebo run-in period, 1,701 patients were randomized to receive placebo (N=567) or BRENZAVVY 20 mg (N=1,134) orally once daily.
At baseline, nearly all patients (99.4%) were treated with one or more antidiabetic medications including metformin (77%), insulin (53%), sulfonylureas (40%), DPP-4 inhibitors (13%) and thiazolidinediones (3%). The mean age of the population was 64 years of age and 11% of the patients were older than 75 years of age. Seventy percent (70%) were male and 77% were White, 10% were Asian, 9% were American Indian or Alaskan Native, and 4% were Black. Fifteen percent (15%) were Hispanic/Latino. At baseline, the mean duration of diabetes was 15 years and the mean HbA1c was 8.3%. The mean baseline eGFR was 78 mL/min/1.73 m²; 80% of patients had an eGFR >60 mL/min/1.73 m² and 20% of patients had an eGFR 45 to 60 mL/min/1.73 m².
Glycemic Response
Treatment with BRENZAVVY provided a statistically significant reduction in HbA1c at Week 24 compared to treatment with placebo (see Table 10). In addition, BRENZAVVY provided a statistically significant reduction in HbA1c compared to placebo in a subset of patients using background insulin (N=902, difference from placebo -0.5% [95% CI: -0.6, -0.4]) and in a subset of patients using background sulfonylureas (N=313, difference from placebo -0.4% [95% CI:-0.6, -0.2]).
Table 10. Glycemic Results from a 24-Week Trial in Adults with Type 2 Diabetes Mellitus with Established CVD or Multiple CVD Risk Factors (Trial 6):
| Placebo N=567 | BRENZAVVY N=1133 | |
|---|---|---|
| HbA1c (%) | ||
| Baseline mean | 8.3 | 8.3 |
| Change from baseline [adjusted mean (SE)]a | -0.4 (0.04) | -0.8 (0.03) |
| Difference from placebo [adjusted mean] (95% CI) | -0.4 (-0.5, -0.4)* | |
| Proportion of patients (%) achieving HbA1c <7%b | 17% | 29% |
| FPG (mg/dL) | ||
| Baseline mean | 162 | 166 |
| Change from baseline [adjusted mean (SE)]c | -4 (2) | -23 (1) |
| Difference from placebo [adjusted mean] (95% CI) | -20 (-24, -15) | |
SE: Standard Error; CI: Confidence Interval; FPG: Fasting plasma glucose
* Statistically significant (multiplicity adjusted one-sided p-value <0.025)
a Intention to treat population. ANCOVA was used to analyze data using imputed values by return to baseline analysis for missing data at week 24 (7% and 6% for bexagliflozin and placebo, respectively). The ANCOVA model included treatment, region, baseline eGFR category (<60 or ≥60 mL/min/1.73m²), baseline BMI category (<25 or ≥25 kg/m²), history of heart failure (yes or no), insulin use or not, and baseline HbA1c value.
b Crude proportion using imputed HbA1c values for missing data at week 24 and averaged across multiply imputed datasets
c Same model as for HbA1c endpoint but with baseline FPG instead of baseline HbA1c as a covariate.
Body Weight and Systolic Blood Pressure Results
The mean baseline body weight in a subgroup of patients with baseline BMI ≥25 kg/m² was 95 kg (N=522) and 97 kg (N=1047) in the placebo and BRENZAVVY groups, respectively. The mean changes from baseline to Week 24 were -0.3 kg and -2.7 kg in the placebo and BRENZAVVY groups, respectively. The difference from placebo (95% CI) for BRENZAVVY was -2.3 kg (-2.8, -1.9).
The mean baseline systolic blood pressure (SBP) in a subgroup of patients with baseline values ≥140 mmHg was 150 mmHg in the placebo (N=215) and BRENZAVVY (N=448) groups. The mean changes in SBP from baseline to Week 24 were -6.6 mmHg and -9.2 mmHg in the placebo and BRENZAVVY groups, respectively. The difference from placebo (95% CI) for BRENZAVVY was -2.7 mmHg (-5.2, -0.1).
Major Adverse Cardiovascular Events (MACE) Results
Trial 6 was used to assess the impact of BRENZAVVY on MACE (a composite of cardiovascular death, non-fatal myocardial infarction, nonfatal stroke, and hospitalization for unstable angina). The minimum treatment duration was 52 weeks (median duration 2.4 years). In this trial, the proportion of patients who experienced at least one MACE event was 10.1% (57/567) in the placebo group and 7.9% (89/1132) in the BRENZAVVY group (4.2 MACE events per 100 person-years for placebo and 3.3 MACE events per 100 person-years for BRENZAVVY). No increased risk for MACE was observed in the BRENZAVVY group compared to the control group [estimated hazard ratio of 0.77 (95% CI: 0.56, 1.08)]. The BRENZAVVY group was not superior to the placebo group in reducing MACE.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.