CAPLYTA Capsule Ref.[10130] Active ingredients:
Source: FDA, National Drug Code (US) Revision Year: 2019
12.1. Mechanism of Action
The mechanism of action of lumateperone in the treatment of schizophrenia is unknown. However, the efficacy of lumateperone could be mediated through a combination of antagonist activity at central serotonin 5-HT2A receptors and postsynaptic antagonist activity at central dopamine D2 receptors.
12.2. Pharmacodynamics
Lumateperone has high binding affinity for serotonin 5-HT2A receptors (Ki = 0.54 nM) and moderate binding affinity for dopamine D2 (Ki = 32 nM) receptors. Lumateperone has moderate binding affinity for serotonin transporters (Ki = 33 nM). Lumateperone also has moderate binding affinity for dopamine D1 (41 nM) and D4 and adrenergic alpha1A and alpha1B receptors (Ki projected at <100 nM) but has low binding affinity (less than 50% inhibition at 100 nM) for muscarinic and histaminergic receptors.
Cardiac Electrophysiology
QTcF interval was evaluated in a randomized, placebo- and active- (moxifloxacin 400 mg) controlled, four-arm crossover study utilizing concentration-QTc effect modeling in 33 patients with schizophrenia. The placebo-corrected change from baseline QTcF (90% two-sided upper confidence interval) values of 4.9 (8.9) and 15.8 (19.8) ms for the 42 mg and the supratherapeutic dose of 126 mg (three times the recommended daily dosage) CAPLYTA, respectively, administered orally once daily for 5 days.
12.3. Pharmacokinetics
Following once daily oral administration of CAPLYTA, lumateperone steady state is reached in about 5 days. Increase in steady-state exposure is approximately dose-proportional in the range of 21 mg to 56 mg. A large inter-subject variability in lumateperone PK parameters was observed, with coefficients of variation for Cmax (peak plasma concentration) and AUC (area under the concentration vs time curve) ranging from 68% to 97% at steady state.
Absorption
The absolute bioavailability of lumateperone capsules is about 4.4%. Cmax of lumateperone is reached approximately 1-2 hours after CAPLYTA dosing.
Effect of Food
Ingestion of a high-fat meal with CAPLYTA lowers lumateperone mean Cmax by 33% and increases mean AUC by 9%. Median Tmax was delayed about 1 hour (from 1 hour at fasted state to 2 hours in the presence of food).
Distribution
Protein binding of lumateperone is 97.4% at 5 µM (about 70-fold higher than therapeutic concentrations) in human plasma. The volume of distribution of lumateperone following intravenous administration is about 4.1 L/kg.
Elimination
The clearance of lumateperone is approximately 27.9 L/hour and the terminal half-life is about 18 hours after intravenous administration.
Metabolism
Lumateperone is extensively metabolized with more than twenty metabolites identified in vivo. After a single 14C-labeled oral dose, lumateperone and glucuronidated metabolites represent about 2.8% and 51% of the total plasma radioactivity, respectively. In vitro studies show that multiple enzymes, including but not limited to uridine 5'-diphospho-glucuronosyltransferases (UDP-glucuronosyltransferase, UGT) 1A1, 1A4, and 2B15, aldoketoreductase (AKR)1C1, 1B10, and 1C4, and cytochrome P450 (CYP) 3A4, 2C8, and 1A2, are involved in the metabolism of lumateperone.
Excretion
In a human mass‑balance study, 58% and 29% of the radioactive dose was recovered in the urine and feces, respectively. Less than 1% of the dose was excreted as unchanged lumateperone in the urine.
Specific Populations
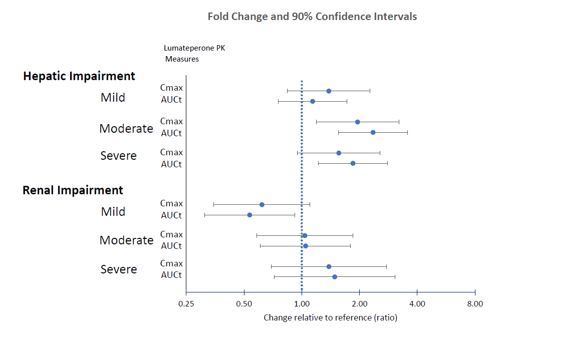
Effects of hepatic or renal impairment on lumateperone exposure are presented in Figure 1. No clinically significant differences in the pharmacokinetics of lumateperone were observed based on age, sex, or race.
Figure 1. Effects of Intrinsic Factors on Lumateperone Pharmacokinetics:
Drug Interaction Studies
Clinical Studies
The effects of other drugs on the exposures of lumateperone are presented in Figure 2.
Figure 2. Effects of Other Drugs on Lumateperone Pharmacokinetics:
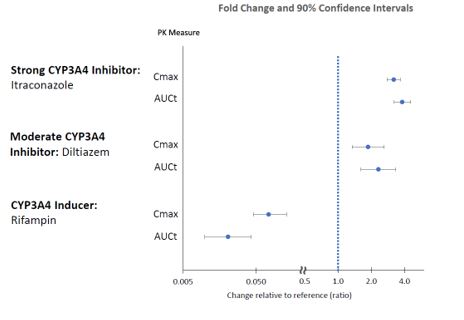
CYP3A4 substrates: No clinically significant differences in the pharmacokinetics of midazolam (CYP3A4 substrate) or its metabolite 1-hydroxymidazolam were observed when used concomitantly with single or multiple doses of lumateperone in patients with schizophrenia.
In Vitro Studies
Lumateperone showed little to no inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5. It showed no induction of CYP1A2, CYP2B6, or CYP3A4.
Lumateperone did not appear to be a P-gp or BCRP substrate. It showed little to no inhibition of OCT2, OAT1, OAT3, OATP1B3, or OATP1B1.
13.1. Carcinogensis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies were conducted in rats and mice, and results showed no carcinogenic potential in either species.
In Sprague-Dawley rats, males were administered lumateperone (free base) at oral doses of 3.5, 7 or 14 mg/kg/day and females were administered lumateperone at oral doses of 3.5, 10.5, or 21 mg/kg/day for the first 385 days, then doses were reduced for the two higher dose groups so that the females were administered 3.5, 7 or 14 mg/kg/day, respectively, for the duration of the study. In this study the no adverse effect level for neoplastic lesions was determined to be 14 mg/kg/day (84 mg/m²/day) for males and 10.5/7 mg/kg/day (42 mg/m²/day) for females, which are 1.6 times (females) to 3.2 times (males) the MRHD on a mg/m² basis.
Male and female CD-1 mice were administered lumateperone at oral doses of 3.5, 10.5 or 21 mg/kg/day for the first 35 days, then doses were reduced to 1.4, 4.9, and 14 mg/kg/day, respectively, for the duration of the study. In this study, the no adverse effect level for neoplastic lesions was determined to be 10.5/4.9 mg/kg/day (15 mg/m²/day) for each sex which is 0.6 times the MRHD on a mg/m² basis.
Mutagenesis
No evidence of mutagenic potential was found in the in vitro bacterial reverse mutation assay (Ames test) and the mouse lymphoma test without metabolic activation. Lumateperone was positive in the Ames test only in the presence of metabolic activation and only in the TA1537 strain and was positive in the mouse lymphoma test only in the presence of metabolic activation and only at high concentrations that inhibited cell growth; together these results were thought to be related to solubility limits and/or nonspecific effects on cellular function. Lumateperone was negative for clastogenic activity in the in vivo micronucleus assay in rats and was not genotoxic in the in vivo Comet assay in rats.
Impairment of Fertility
Female rats were treated with oral doses of 3.5, 10.5, 21 or 42 mg/kg/day lumateperone (free base) (0.8, 2.4, 4.9, and 9.7 times the MRHD on a mg/m² basis) prior to mating and continuing through conception and implantation. Estrus cycle irregularities were observed at doses ≥10.5 mg/kg/day. Decreases in the median number of corpora lutea and implantation sites, and increases in the number of non-gravid uteruses, were recorded at 42 mg/kg/day. Decreased gestation body weight and body weight gain, and increases in time to mating, were observed at 21 and 42 mg/kg/day.
Male rats were treated with oral doses of 3.5, 10.5, 21 or 42 mg/kg/day lumateperone (0.8, 2.4, 4.9, and 9.7 times the MRHD on a mg/m² basis) for 9 weeks prior to mating and throughout 14 days of mating. Decreased sperm motility, changes in sperm morphology, reduced epididymal counts, and adverse histopathology changes in testes and epididymides were observed at 21 and 42 mg/kg/day.
13.2. Animal Toxicology and/or Pharmacology
Oral administration of lumateperone caused systemic intracytoplasmic accumulation of pigmented material in dogs, rats, and mice at clinically relevant exposures (AUC). Intracytoplasmic pigmentation appeared to be localized in lysosomes. Accumulation of pigmented material persisted without reversal at the end of 1- to 2-month drug-free periods. Pigmented material was observed in the brain and spinal cord of all three species, and in the heart and eye of rats. Although the composition of the pigmented material was not established, the material is likely polymers or protein adducts formed from aniline metabolites of lumateperone.
In the dog, accumulation of pigmented material in the brain and spinal cord was associated with neuronal degeneration and necrosis, followed by axonal degeneration and histiocytic inflammation after oral administration of lumateperone for up to 9 months. In the rat, accumulation of pigmented material was associated with degenerative changes and signs of an inflammatory response in the spinal cord, peripheral nervous system, eye, and heart after oral administration of lumateperone for up to 2 years. Although overt degenerative changes were not observed in the rat brain, the presence of pigment-containing infiltrating macrophages is consistent with an inflammatory response.
The role of intracytoplasmic pigmented material in causing these lesions was not definitively established; however, the colocalization of pigmented material in tissues with degenerative changes and signs of inflammation is supportive. Alternatively, the aniline metabolites of lumateperone may undergo metabolic activation forming reactive metabolites that contribute to the observed toxicities. The role of intracellular accumulation of lumateperone or its non-aniline metabolites in these toxicities could not be ruled out.
The aniline metabolites thought to be responsible for these toxicities were detected in dogs and rats but were not present in humans at quantifiable levels. Based on all the available evidence, these toxicities do not appear to be relevant to humans.
14. Clinical Studies
CAPLYTA was evaluated for the treatment of schizophrenia in two placebo-controlled trials.
Study 1 (NCT01499563) was a four-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients with a diagnosis of schizophrenia according to the DSM-IV-TR criteria. The primary efficacy measure was the Positive and Negative Syndrome Scale (PANSS) total score. The PANSS is a 30-item scale used to measure symptoms of schizophrenia. Each item is rated by a clinician on a seven-point scale. A score of 1 indicates the absence of symptoms, and a score of 7 indicates extremely severe symptoms. The PANSS total score may range from 30 to 210 with higher scores reflecting greater overall symptom severity.
A total of 335 patients were randomized to receive CAPLYTA 42 mg, CAPLYTA 84 mg (two times the recommended daily dose), an active comparator, or placebo. The study was not designed to allow for efficacy comparison of CAPLYTA and the active comparator. Demographic and baseline disease characteristics were similar for the CAPLYTA, active comparator, and placebo groups. Median age was 42 years (range 20 to 55 years). 17% were female, 19% were Caucasian, and 78% were African American.
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant reduction from baseline to Day 28 in the PANSS total score. The treatment effect in the CAPLYTA 84 mg group (vs. placebo) was not statistically significant. The results of Study 1 are shown in Table 3.
Study 2 (NCT02282761) was a four-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients with a diagnosis of schizophrenia according to the DSM-5 criteria. The primary efficacy measure was the PANSS total score.
A total of 450 patients were randomized to receive CAPLYTA 28 mg (two-thirds the recommended daily dose), CAPLYTA 42 mg, or placebo. Demographic and baseline disease characteristics were similar for the CAPLYTA and placebo groups. Median age was 44 years (range 19 to 60 years); 23% were female, 26% were Caucasian and 66% were African American.
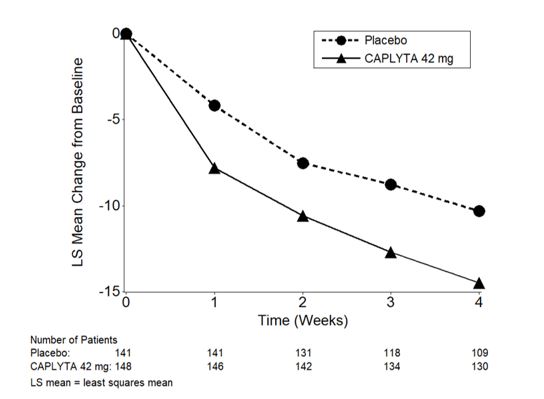
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant reduction from baseline to Day 28 in the PANSS total score. The treatment effect in the CAPLYTA 28 mg group (vs. placebo) was not statistically significant. The results of Study 2 are shown in Table 3.
Studies 1 and 2 did not include any patients aged 65 or older. Examination of subgroups by sex and race did not suggest differences in response in either study.
Table 3. Primary Efficacy Results for Change from Baseline in PANSS Total Score in Patients with Schizophrenia (Studies 1 and 2):
| Primary Efficacy Endpoint: PANSS Total Score | |||||
|---|---|---|---|---|---|
| Study Number | Treatment Group | N | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference (95% CI) |
| 1 | CAPLYTA (42 mg)* | 84 | 88.1 (11.0) | -13.2 (1.7) | -5.8 (-10.5, -1.1)a |
| Placebo | 85 | 86.3 (13.1) | -7.4 (1.7) | -- | |
| 2 | CAPLYTA (42 mg)* | 150 | 90.0 (9.6) | -14.5 (1.3) | -4.2 (-7.8, -0.6) |
| Placebo | 150 | 89.0 (10.3) | -10.3 (1.3) | -- | |
The PANSS total score may range from 30 to 210; higher scores reflect greater symptom severity.
SD: standard deviation; SE: standard error; LS Mean: least squares mean; CI: unadjusted confidence interval.
a Difference (drug minus placebo) in LS mean change from baseline not adjusted for sample size increase after unblinded interim analysis.
* Statistically significantly superior to placebo.
Figure 3. Change from Baseline in PANSS Total Score by Time (Weeks) in Patients with Schizophrenia in Study 2:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.