CASGEVY Dispersion for infusion Ref.[108594] Active ingredients: Exagamglogene autotemcel
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Vertex Pharmaceuticals (Ireland) Limited, Unit 49, Block F2, Northwood Court, Santry, Dublin 9, D09 T665, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other haematological agents, other haematological agents
ATC code: B06AX05
Mechanism of action
Casgevy is a cell therapy consisting of autologous CD34+ HSPCs ex vivo edited by CRISPR/Cas9-technology. The highly specific guide RNA enables CRISPR/Cas9 to make a precise DNA double-strand break at the critical transcription factor binding site (GATA1) in the erythroid specific enhancer region of the BCL11A gene. As a result of the editing, GATA1 binding is irreversibly disrupted and BCL11A expression reduced. Reduced BCL11A expression results in an increase in γ-globin expression and foetal haemoglobin (HbF) protein production in erythroid cells, addressing the absent globin in transfusion-dependent β-thalassemia (TDT) and the aberrant globin in sickle cell disease (SCD), which are the underlying causes of disease. In patients with TDT, γ-globin production is expected to correct the α-globin to non-α-globin imbalance, thereby reducing ineffective erythropoiesis and haemolysis and increasing total haemoglobin levels. In patients with severe SCD, HbF expression is expected to reduce intracellular HbS concentration, preventing the red blood cells from sickling.
Clinical efficacy and safety
The efficacy of Casgevy was evaluated in adolescent and adult patients with transfusion-dependent β-thalassemia (TDT) or sickle cell disease (SCD) in two open-label, single-arm studies (study 111 and study 121) and one long-term follow-up study (study 131).
Transfusion-dependent β-thalassemia
Study 111 is an ongoing open-label, multicentre, single-arm study to evaluate the safety and efficacy of Casgevy in adult and adolescent patients with transfusion-dependent β-thalassemia. After completion of 24 months of follow-up in study 111, patients were invited to enrol in study 131, an ongoing long-term safety and efficacy study.
Patients were eligible for the study if they had a history of requiring at least 100 mL/kg/year or 10 units/year of RBC transfusions in the 2 years prior to enrolment. Patients were also required to have a Lansky or Karnofsky performance score of ≥ 80%.
Patients were excluded from the study if they had an available HLA-matched related HSC donor. Patients who had severely elevated iron in the heart (i.e., patients with cardiac T2* less than 10 msec by magnetic resonance imaging [MRI]), or advanced liver disease were excluded from the study. MRI of the liver was performed on all patients. Patients with MRI results demonstrating liver iron content ≥15 mg/g underwent liver biopsy for further evaluation. Patients with a liver biopsy demonstrating bridging fibrosis or cirrhosis were excluded.
Of the 59 patients who initiated mobilisation in study 111, 3 patients (5.1%) discontinued prior to Casgevy infusion, all due to withdrawn consent.
The key demographics and baseline characteristics are shown in Table 5 for (1) all patients enrolled in study 111 and (2) all patients infused with Casgevy in study 111.
Table 5. Study 111 demographics and baseline characteristics:
| Demographics and disease characteristics | Casgevy Enrolled patients (N=59)§ | Casgevy Infused patients† (N=54) |
|---|---|---|
| Age, n (%) | ||
| Adults (≥18 and ≤35 years) | 39 (66.1%) | 35 (64.8%) |
| Adolescents (≥12 and <18 years) | 20 (33.9%) | 19 (35.2%) |
| All ages (≥12 and ≤35 years) Median (min, max) | 19 (12, 35) | 20 (12, 35) |
| Sex, n (%) | ||
| Female | 28 (47.5%) | 25 (46.3%) |
| Male | 31 (52.5%) | 29 (53.7%) |
| Race, n (%) | ||
| Asian | 25 (42.4%) | 23 (42.6%) |
| White | 19 (32.2%) | 18 (33.3%) |
| Multiracial | 3 (5.1%) | 3 (5.6%) |
| Other | 3 (5.1%) | 2 (3.7%) |
| Not collected | 9 (15.3%) | 8 (14.8%) |
| Genotype, n (%) | ||
| β0/β0-like‡ | 38 (64.4%) | 33 (61.1%) |
| Non-β0/β0-like | 21 (35.6%) | 21 (38.9%) |
| Baseline annualised RBC transfusion volume (mL/kg) | ||
| Median (min, max) | 1211.2 (48.3, 330.9) | 1205.7 (48.3, 330.9) |
| Baseline annualised RBC transfusion episodes | ||
| Median (min, max) | 16.5 (5.0, 34.5) | 16.5 (5.0, 34.5) |
| Spleen intact, n (%) | 43 (72.9%) | 38 (70.4%) |
| Baseline liver iron concentration (mg/g) | ||
| Median (min, max) | 3.5 (1.2, 14.8) | 3.5 (1.2, 14.0) |
| Baseline cardiac iron T2* (msec) | ||
| Median (min, max) | 34.1 (12.4, 61.1) | 34.4 (12.4, 61.1) |
| Baseline serum ferritin (pmol/L) | ||
| Median (min, max) | 3100.9 (584.2, 10837.3) | 3115.5 (584.2, 10837.3) |
§ N represents the total number of enrolled patients who signed informed consent.
† Interim analysis conducted based on April 2023 data cut with 54 patients administered Casgevy and 2 patients awaiting Casgevy infusion.
‡ Low to no endogenous β-globin production (β0/β0, β0/IVS-I-110 and IVS-I-110/IVS-I-110).
Mobilisation and apheresis
To maintain a total Hb concentration ≥ 11 g/dL patients underwent RBC transfusions prior to mobilisation and apheresis and continued receiving transfusions until the initiation of myeloablative conditioning.
To mobilise stem cells for apheresis, patients in study 111 were administered granulocyte-colony stimulating factor (G-CSF). Patients with a spleen were administered a planned dose of 5 mcg/kg G-CSF approximately every 12 hours via intravenous or subcutaneous injection for 5 to 6 days. Splenectomised patients were administered a planned dose of 5 mcg/kg G-CSF once daily for 5 to 6 days. The dose was increased to every 12 hours in splenectomised patients if there was no increase in white blood cell (WBC) or peripheral blood CD34+ counts. After 4 days of G-CSF administration, all patients received plerixafor at a planned dose of 0.24 mg/kg administered via subcutaneous injection approximately 4- to 6 hours prior to each planned apheresis. Apheresis was carried out for up to 3 consecutive days to achieve the target collection of cells for manufacture and for the unmodified rescue CD34+ cells. The mean (SD) and median (min, max) number of mobilisation and apheresis cycles required for manufacture Casgevy and for the collection of rescue CD34+ cells were 1.3 (0.7) and 1 (1, 4), respectively.
Pre-treatment conditioning
All patients received full myeloablative conditioning with busulfan prior to treatment with Casgevy. Busulfan was administered for 4 consecutive days intravenously via a central venous catheter at a planned starting dose of 3.2 mg/kg/day once daily or 0.8 mg/kg every 6 hours. Busulfan plasma levels were measured by serial blood sampling and the dose adjusted to maintain exposure in the target range. For once daily dosing, four-day target cumulative busulfan exposure was 82 mg*h/L (range 74 to 90 mg*h/L), corresponding to AUC0-24h of 5000 μM*min (range: 4500 to 5500 μM*min). For dosing every 6 hours, the four-day target cumulative busulfan exposure was 74 mg*h/L (range 59 to 89 mg*h/L), corresponding to AUC0-6h of 1125 μM*min (range: 900 to 1350 μM*min).
All patients received anti-seizure prophylaxis with agents other than phenytoin prior to initiating busulfan conditioning. Phenytoin was not used for anti-seizure prophylaxis because of its induction of cytochrome P450 and resultant increased clearance of busulfan.
Prophylaxis for hepatic venoocclusive disease (VOD)/hepatic sinusoidal obstruction syndrome was administered per institutional guidelines.
Casgevy administration
Patients were administered Casgevy with a median (min, max) dose of 8.0 (3.0, 19.7) × 106 cells/kg as an intravenous infusion. All patients were administered an antihistamine and an antipyretic prior to Casgevy infusion.
After Casgevy administration
G-CSF was not recommended within the first 21 days after Casgevy infusion. As Casgevy is an autologous therapy, immunosuppressive agents were not required after initial myeloablative conditioning.
Efficacy results – β-thalassemia
An interim analysis (IA) was conducted with 42 patients administered Casgevy and eligible for the primary efficacy analysis. The primary efficacy set (PES) was defined as all subjects who had been followed for at least 16 months after Casgevy infusion. At the time of the interim analysis 59 patients were enrolled and 54 patients had been administered Casgevy. The median (min, max) total duration of follow-up was 22.8 (2.1, 51.1) months from the time of Casgevy infusion.
The efficacy of Casgevy was assessed based on evaluation of patients with at least 16 months of follow-up. The primary endpoint was the proportion of patients achieving transfusion independence for 12 consecutive months (TI12), defined as maintaining weighted average Hb ≥9 g/dL without RBC transfusions for at least 12 consecutive months any time within the first 24 months after Casgevy infusion in study 111, evaluated starting 60 days after the last RBC transfusion for post-transplant support or TDT disease management.
Efficacy data are presented in Table 6 and Table 7. Table 6 shows the primary endpoint for (1) all patients enrolled in study 111 and (2) all patients infused with Casgevy in study 111. Table 7 shows secondary endpoints for patients infused with Casgevy in study 111.
Table 6. Primary efficacy outcome in patients with TDT:
| Primary endpoint | Casgevy Enrolled patients* (N=45)† | Casgevy Infused patients* (N=42)‡ |
|---|---|---|
| Proportion of patients achieving TI12§ | ||
| n (%) | 39 (86.7%) | 39 (92.9%) |
| (95% CI) | (73.2%, 94.9%) | (80.5%, 98.5%) |
* Interim analysis conducted based on April 2023 data cut.
† N represents the total number of enrolled patients who signed inform consent, and excludes patients who were waiting to receive Casgevy at the time of the analysis, or patients who were not yet evaluable for the primary efficacy endpoint.
‡ N represents the total number of patients in the primary efficacy set (PES), a subset of the full analysis set (FAS). The PES was defined as all subjects who had been infused with Casgevy and had been followed for at least 16 months after Casgevy infusion. Patients who had less than 16 months of follow-up due to death, or discontinuation due to Casgevy-related adverse events, or continuously received RBC transfusions for more than 12 months after Casgevy infusion were also included in this set.
§ TI12 is defined as maintaining weighted average Hb ≥9 g/dL without RBC transfusions for at least 12 consecutive months any time after Casgevy infusion. The evaluation of TI12 starts 60 days after last RBC transfusion for post-transplant support or TDT disease management.
Table 7. Secondary efficacy outcomes in patients with TDT:
| Secondary endpoints | Casgevy Infused patients* (N=42)† |
|---|---|
| Duration of transfusion-independent period in patients who have achieved TI12 (months) | |
| n | 39 |
| Median (min, max) | 22.3 (13.5, 48.1) |
| Total Hb (g/dL) | |
| at Month 6 | |
| n | 42 |
| Mean (SD) | 12.1 (2.0) |
| at Month 24 | |
| n | 23 |
| Mean (SD) | 12.9 (2.4) |
| HbF (g/dL) | |
| at Month 6 | |
| n | 42 |
| Mean (SD) | 10.8 (2.8) |
| at Month 24 | |
| n | 23 |
| Mean (SD) | 11.5 (2.7) |
* Interim analysis conducted based on April 2023 data cut.
† N represents the total number of patients in the primary efficacy set (PES), a subset of the full analysis set (FAS). The PES was defined as all patients who had been infused with Casgevy and had been followed for at least 16 months after Casgevy infusion. Subjects who had less than 16 months follow-up due to death or discontinuation due to Casgevy-related adverse events, or continuously received RBC transfusions for more than 12 months after Casgevy infusion were also included in this set.
SD: Standard Deviation
All patients who achieved TI12 remained transfusion-independent, with a median (min, max) duration of transfusion-independence of 22.3 (13.5, 48.1) months and normal weighted average total Hb levels (mean [SD] 13.2 [1.4] g/dL). The median (min, max) time to last RBC transfusion for patients who achieved TI12 was 28 (11, 91) days following Casgevy infusion. Three patients did not achieve TI12. These patients had reductions in frequency of RBC transfusions over time, and then stopped receiving transfusions between 12.2 and 21.6 months after Casgevy infusion, consistent with an overall slower haematopoietic recovery.
Total Hb (g/dL) and HbF (g/dL) levels over time are provided in Figure 1 for all patients administered Casgevy for the treatment of β-thalassemia.
Figure 1. Mean total Hb (g/dL) and HbF (g/dL) levels over time in patients with TDT:

Mean values are plotted in the line, mean +Standard Error (SE) and mean -SE values are plotted as bars at each visit. The numbers of patients with values available at the corresponding visits are shown beneath the figure.
Increases in mean (SD) total Hb and HbF levels were observed as early as month 3 after Casgevy infusion and continued to increase to 12.2 (2.0) g/dL and 10.9 (2.7) g/dL respectively at month 6. After month 6, levels of total Hb and HbF were maintained thereafter, with HbF comprising ≥88% of total Hb.
All patients who achieved TI12 in Study 111 (n=39) had normal (28/39 patients, 71.8%) or near-normal (11/39 patients, 28.2%) weighted average total Hb levels. The patients with near-normal weighted average total Hb levels included 6 males and 5 females, with weighted average total Hb within <0.1 to 0.7 g/dL and within <0.4 to 1.4 g/dL of the WHO age and sex dependent reference threshold, respectively.
Subgroup analyses evaluating the effects in age, sex, race or genotype subgroups on transfusion-related endpoints and haematological parameters did not suggest differences due to these factors.
Sickle cell disease
Study 121 is an ongoing open-label, multicentre, single-arm study to evaluate the safety and efficacy of Casgevy in adult and adolescent patients with severe sickle cell disease. After completion of 24 months of follow-up in study 121, patients were invited to enrol in study 131, an ongoing long-term safety and efficacy study.
Patients were eligible for the study if they had a history of at least 2 severe vaso-occlusive crisis (VOC) events per year in the 2 years prior to screening, which were defined as:
- Acute pain event requiring a visit to a medical facility and administration of pain medications (opioids or intravenous non-steroidal anti-inflammatory drugs [NSAIDs]) or RBC transfusions
- Acute chest syndrome
- Priapism lasting >2 hours and requiring a visit to a medical facility
- Splenic sequestration.
Patients with HbS/S, HbS/β0 and HbS/β+ genotypes were eligible for inclusion. Patients were also required to have a Lansky or Karnofsky performance score of ≥80%.
Patients were excluded from the study if they had an available HLA-matched related HSC donor. Patients were excluded if they had advanced liver disease, history of untreated Moyamoya disease, or presence of Moyamoya disease that in the opinion of the investigator put the patient at risk of bleeding. Patients aged 12 to 16 years were required to have normal transcranial doppler (TCD) and patients aged 12 to 18 years were excluded if they had any history of TCD in the middle cerebral artery and the internal carotid artery.
Of the 58 patients that initiated mobilisation in study 121, 11 patients (19.0%) discontinued after starting mobilisation and apheresis and prior to Casgevy administration. Six patients (10.3%) did not achieve the minimum dose. Five patients (8.6%) discontinued due to noncompliance, withdrawn consent, or no longer meeting eligibility requirements.
The key demographics and baseline characteristics are shown in Table 8, below, for (1) all patients enrolled in study 121 and (2) all patients infused with Casgevy in study 121.
Table 8. Study 121 demographics and baseline characteristics:
| Demographics and disease characteristics | Casgevy Enrolled patients (N=63)* | Casgevy Infused patients (N=43)† |
|---|---|---|
| Age (years), n (%) | ||
| Adults (≥18 and ≤35 years) | 50 (79.4%) | 31 (72.1%) |
| Adolescents (≥12 and <18 years) | 13 (20.6%) | 12 (27.9%) |
| All ages (≥12 and ≤35 years) Median (min, max) | 21.0 (12, 35) | 20 (12, 34) |
| Sex, n (%) | ||
| Male | 36 (57.1%) | 24 (55.8%) |
| Female | 27 (42.9%) | 19 (44.2%) |
| Race, n (%) | ||
| Black or African American | 55 (87.3%) | 37 (86.0%) |
| White | 4 (6.3%) | 3 (7.0%) |
| Other | 4 (6.3%) | 3 (7.0%) |
| Genotype, n (%)‡ | ||
| βS/βS | 58 (92.1%) | 39 (90.7%) |
| βS/β0 | 3 (4.8%) | 3 (7.0%) |
| βS/β+ | 2 (3.2%) | 1 (2.3%) |
| Annualised rate of severe VOCs in the 2 years prior to enrolment (events/year) | ||
| Median (min, max) | 3.5 (2.0, 19.0) | 3.5 (2.0, 18.5) |
| Annualised rate of hospitalisations due to severe VOCs in the 2 years prior to enrolment (events/year) | ||
| Median (min, max) | 2.5 (0.0, 11.0) | 2.5 (0.5, 9.5) |
| Annualised duration of hospitalisation due to severe VOCs in the 2 years prior to enrolment (days/year) | ||
| Median (min, max) | 15.5 (0.0, 136.5) | 13.5 (2.0, 136.5) |
| Annualised units of RBCs transfused for SCD-related indications in the 2 years prior to enrolment (units/year) | ||
| Median (min, max) | 5.0 (0.0, 86.1) | 5.0 (0.0, 86.1) |
* N represents the total number of enrolled patients who signed inform consent.
† Interim analysis conducted based on April 2023 data cut with 43 patients having been administered Casgevy and 4 patients awaiting Casgevy infusion.
‡ There is no data available for patients with other genotypes.
Mobilisation and apheresis
Patients underwent red blood cell exchange or simple transfusions for a minimum of 8 weeks before the planned start of mobilisation and continued receiving transfusions or red blood cell exchanges until the initiation of myeloablative conditioning. HbS levels were maintained at <30% of total Hb while keeping total Hb concentration ≤11 g/dL.
To mobilise stem cells for apheresis, patients in study 121 were administered plerixafor at a planned dose of 0.24 mg/kg via subcutaneous injection approximately 2 to 3 hours prior to each planned apheresis. Patients underwent apheresis for up to 3 consecutive days to achieve the target collection of cells for manufacture and for the unmodified rescue CD34+ cells. The median (min, max) and mean (SD) number of mobilisation and apheresis cycles required for manufacture Casgevy and for the collection of rescue CD34+ cells were 2 (1, 6) and 2.21 (1.30), respectively.
Pre-treatment conditioning
All patients received full myeloablative conditioning with busulfan prior to receiving Casgevy. Busulfan was administered for 4 consecutive days intravenously via a central venous catheter at a planned starting dose of 3.2 mg/kg/day once daily or 0.8 mg/kg every 6 hours. Busulfan plasma levels were measured by serial blood sampling and the dose adjusted to maintain exposure in the target range. For once daily dosing, the four-day target cumulative busulfan exposure of 82 mg*h/L (range 74 to 90 mg*h/L), corresponding to AUC0-24h of 5000 μM*min (range: 4500 to 5500 μM*min). For dosing every 6 hours, the four-day target cumulative busulfan exposure of 74 mg*h/L (range 59 to 89 mg*h/L), corresponding to AUC0-6h of 1125 μM*min (range 900 to 1350 μM*min).
All patients received anti-seizure prophylaxis with agents other than phenytoin prior to initiating busulfan conditioning. Phenytoin was not used for anti-seizure prophylaxis because of its induction of cytochrome P-450 and resultant increased clearance of busulfan.
Prophylaxis for hepatic veno-occlusive disease (VOD)/hepatic sinusoidal obstruction syndrome was administered, per regional and institutional guidelines.
Casgevy administration
Patients were administered Casgevy with a median (min, max) dose of 4.0 (2.9 to 14.4) × 106 cells/kg as an intravenous infusion. All patients were administered an antihistamine and an antipyretic prior to Casgevy infusion.
After Casgevy administration
G-CSF was not recommended within the first 21 days after Casgevy infusion. As Casgevy is an autologous therapy, immunosuppressive agents were not required after initial myeloablative conditioning.
Efficacy results – sickle cell disease
An interim analysis was conducted with 29 patients administered Casgevy and eligible for the primary efficacy analysis. The primary efficacy set (PES) was defined as all patients who had been followed for at least 16 months after Casgevy infusion. At the time of the interim analysis 63 patients were enrolled and 43 patients had been administered Casgevy. The median (min, max) total duration of follow-up was 17.5 (1.2, 46.2) months from the time of Casgevy infusion.
The efficacy of Casgevy was based on evaluation of patients with at least 16 months of follow-up. The primary endpoint was the proportion of patients who did not experience any severe VOCs for at least 12 consecutive months any time within the first 24 months after Casgevy infusion in study 121 (VF12, primary efficacy endpoint). For this endpoint, a severe VOC was defined as either (a) an acute pain event requiring a visit to a medical facility and administration of pain medications (opioids or intravenous non-steroidal anti-inflammatory drugs [NSAIDs]) or RBC transfusions), (b) acute chest syndrome, (c) priapism lasting >2 hours and requiring a visit to a medical facility, or (d) splenic sequestration. The proportion of patients who did not require hospitalisation due to severe VOCs for at least 12 consecutive months (HF12, key secondary endpoint) was also assessed. The evaluation of VF12 and HF12 began 60 days after last RBC transfusion for post-transplant support or SCD management.
Efficacy data are presented in Table 9 and Table 10. Table 9 shows the primary endpoint for (1) all patients enrolled in study 121 and (2) all patients infused with Casgevy in study 121. Table 10 shows secondary endpoints for all patients infused with Casgevy in study 121.
Table 9. Primary efficacy outcome in patients with SCD:
| Primary endpoint | Casgevy Enrolled patients* (N=46)† | Casgevy Infused patients* (N=29)‡ |
|---|---|---|
| Proportion of patients achieving VF12 (%)§ | ||
| n (%) | 28 (60.9%) | 28 (96.6%) |
| (95% CI) | (45.4%, 74.9%) | (82.2%, 99.9%) |
* Interim analysis conducted based on April 2023 data cut
† N represents the total number of enrolled patients who signed informed consent, and excludes patients who were waiting to receive Casgevy at the time of the interim analysis, or patients who were dosed but not yet evaluable for the primary efficacy endpoint.
‡ N represents the total number of patients in the primary efficacy set (PES), a subset of the full analysis set (FAS). The PES was defined as all patients who had been infused with Casgevy and had been followed for at least 16 months after Casgevy infusion. Subjects who had less than 16 months follow-up due to death, or discontinuation due to Casgevy-related adverse events, or continuously received RBC transfusions for more than 12 months after Casgevy were also included in this set.
§ VF12 is defined as no severe VOCs for at least 12 consecutive months after Casgevy infusion. The evaluation of VF12 starts 60 days after last RBC transfusion for post-transplant support or SCD management.
Table 10. Secondary efficacy outcomes in patients with SCD:
| Secondary endpoints | Casgevy Infused patients* (N=29)† |
|---|---|
| Proportion of patients free from hospitalisation due to severe VOCs for at least 12 months (HF12) (%)‡ | |
| n (%) | 29 (100%) |
| (95% CI) | (88.1%, 100.0%) |
| Duration of severe VOC-free period in patients who have achieved VF12 (months) | |
| n | 28 |
| Median (min, max) | 20.5 (13.5, 43.6) |
| Proportion of patients with HbF ≥20% at the time of analysis sustained for at least 3, 6 and 12 months (%) | |
| n | 29 |
| % (95% CI) | 100% (88.1%, 100.0%) |
| Total Hb (g/dL) | |
| at Month 6 | |
| n | 27 |
| Mean (SD) | 12.7 (1.7) |
| at Month 24 | |
| n | 15 |
| Mean (SD) | 13.1 (1.9) |
| Proportion of total Hb comprised by HbF (%) | |
| at Month 6 | |
| n | 27 |
| Mean (SD) | 43.1 (6.0) |
| at Month 24 | |
| n | 15 |
| Mean (SD) | 42.2 (5.5) |
* Interim analysis conducted based on April 2023 data cut.
† N represents the total number of patients in the primary efficacy set (PES), a subset of the full analysis set (FAS). The PES was defined as all patients who had been infused with Casgevy and had been followed for at least 16 months after Casgevy infusion. Subjects who had less than 16 months follow-up due to death, or discontinuation due to Casgevy-related adverse events, or continuously received RBC transfusions for more than 12 months after Casgevy were also included in this set.
‡ HF12 is defined as no severe VOC-related inpatient hospitalisations sustained for at least 12 months after Casgevy infusion.
The evaluation of HF12 starts 60 days after last RBC transfusion for post-transplant support or SCD management.
Total Hb (g/dL) and HbF (g/dL) levels over time is provided in Figure 2 for all patients administered Casgevy for the treatment of sickle cell disease.
Figure 2. Mean total Hb and HbF levels over time in patients with SCD:
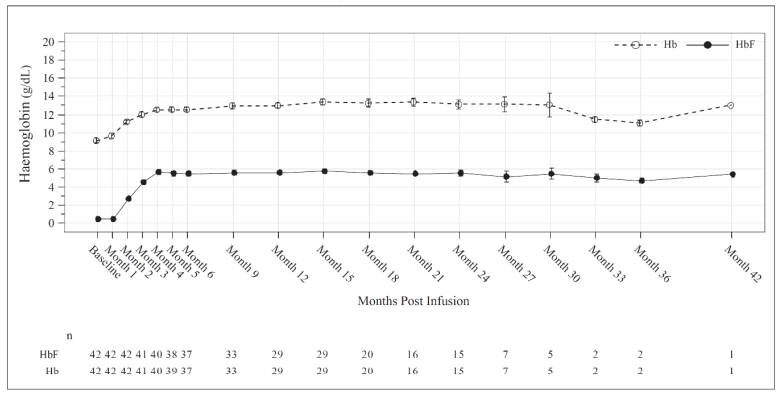
Mean values are plotted in the line, mean +SE and mean -SE values are plotted as bars at each visit. The numbers of patients with values available at the corresponding visits are shown beneath the figure.
Increases in mean (SD) total Hb levels were observed as early as month 3 after Casgevy infusion, continued to increase to 12.5 (1.8) g/dL at month 6 and were maintained thereafter.
The mean (SD) proportion of Hb comprised by HbF was 43.2% (7.6%) at Month 6 and was maintained thereafter.
Consistent with the increase in HbF levels, for all patients dosed the mean (SD) proportion of circulating erythrocytes expressing HbF (F-cells) at month 3 was 70.4% (14.0%) and continued to increase over time to 93.9% (12.6%) at month 6, with levels remaining stable thereafter, indicating sustained pan-cellular expression of HbF.
Subgroup analyses evaluating the effects in age, sex, race or genotype subgroups on VOC-related endpoints and haematological parameters did not suggest differences due to these factors.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Casgevy in one or more subsets of the paediatric population in β-thalassemia and sickle cell disease (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Casgevy is an autologous cellular therapy medicinal product consisting of CD34+ cells that have been edited ex vivo by CRISPR/Cas9. The nature of Casgevy is such that conventional studies on pharmacokinetics, absorption, distribution, metabolism, and elimination are not applicable.
5.3. Preclinical safety data
Casgevy is a CD34+ cell product edited with CRISPR/Cas9 technology; therefore, conventional mutagenicity, carcinogenicity and fertility, reproductive and developmental toxicity studies have not been conducted.
Toxicological characteristics were assessed in sub-lethally irradiated, immunodeficient NSG mice treated with a dose of 3.33 × 107 edited CD34+ cells/kg of body weight. There was no evidence of target organ toxicity or tumorigenicity in the 20-week study.
In vitro studies with exagamglogene autotemcel manufactured from healthy donors and patients showed no evidence of off target editing.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.