CELSENTRI Film-coated tablet Ref.[8785] Active ingredients: Maraviroc
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: ViiV Healthcare BV, Huis ter Heideweg 62, 3705 LZ Zeist, Netherlands
Contraindications
Hypersensitivity to the active substance or to peanut or soya or to any of the excipients listed in section 6.1.
Special warnings and precautions for use
General
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Hepatic disease
The safety and efficacy of maraviroc have not been specifically studied in patients with significant underlying liver disorders.
Cases of hepatotoxicity and hepatic failure with allergic features have been reported in association with maraviroc. In addition, an increase in hepatic adverse reactions with maraviroc was observed during studies of treatment-experienced subjects with HIV infection, although there was no overall increase in ACTG Grade ¾ liver function test abnormalities (see section 4.8). Hepatobiliary disorders reported in treatment-naïve patients were uncommon and balanced between treatment groups (see section 4.8). Patients with pre-existing liver dysfunction, including chronic active hepatitis, can have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice.
Discontinuation of maraviroc should be strongly considered in any patient with signs or symptoms of acute hepatitis, in particular if drug-related hypersensitivity is suspected or with increased liver transaminases combined with rash or other systemic symptoms of potential hypersensitivity (e.g. pruritic rash, eosinophilia or elevated IgE).
There are limited data in patients with hepatitis B and/or C virus co-infection (see section 5.1). Caution should be exercised when treating these patients. In case of concomitant antiviral therapy for hepatitis B and/or C, please refer to the relevant product information for these medicinal products.
There is limited experience in patients with reduced hepatic function, therefore maraviroc should be used with caution in this population (see sections 4.2 and 5.2).
Severe skin and hypersensitivity reactions
Hypersensitivity reactions including severe and potentially life threatening events have been reported in patients taking maraviroc, in most cases concomitantly with other medicinal products associated with these reactions. These reactions included rash, fever, and sometimes organ dysfunction and hepatic failure. Discontinue maraviroc and other suspect agents immediately if signs or symptoms of severe skin or hypersensitivity reactions develop. Clinical status and relevant blood chemistry should be monitored and appropriate symptomatic therapy initiated.
Cardiovascular safety
Limited data exist with the use of maraviroc in patients with severe cardiovascular disease, therefore special caution should be exercised when treating these patients with maraviroc. In the pivotal studies of treatment-experienced patients coronary heart disease events were more common in patients treated with maraviroc than with placebo (11 during 609 PY vs 0 during 111 PY of follow-up). In treatment-naïve patients such events occurred at a similarly low rate with maraviroc and control (efavirenz).
Postural hypotension
When maraviroc was administered in studies with healthy volunteers at doses higher than the recommended dose, cases of symptomatic postural hypotension were seen at a greater frequency than with placebo. Caution should be used when administering maraviroc in patients on concomitant medicinal products known to lower blood pressure. Maraviroc should also be used with caution in patients with severe renal insufficiency and in patients who have risk factors for, or have a history of postural hypotension. Patients with cardiovascular co-morbidities could be at increased risk of cardiovascular adverse reactions triggered by postural hypotension.
Renal impairment
An increased risk of postural hypotension may occur in patients with severe renal insufficiency who are treated with potent CYP3A inhibitors or boosted protease inhibitors (PIs) and maraviroc. This risk is due to potential increases in maraviroc maximum concentrations when maraviroc is co-administered with potent CYP3A inhibitors or boosted PIs in these patients.
Immune reconstitution syndrome
In HIV infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and pneumonia caused by Pneumocystis jiroveci (formerly known as Pneumocystis carinii). Any inflammatory symptoms should be evaluated and treatment initiated when necessary. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occurmany months after initiation of treatment.
Tropism
Maraviroc should only be used when only CCR5-tropic HIV-1 is detectable (i.e. CXCR4 or dual/mixed tropic virus not detected) as determined by an adequately validated and sensitive detection method (see sections 4.1, 4.2 and 5.1). The Monogram Trofile assay was used in the clinical studies of maraviroc. The viral tropism cannot be predicted by treatment history or assessment of stored samples.
Changes in viral tropism occur over time in HIV-1 infected patients. Therefore there is a need to start therapy shortly after a tropism test.
Background resistance to other classes of antiretrovirals have been shown to be similar in previously undetected CXCR4-tropic virus of the minor viral population, as that found in CCR5-tropic virus.
Maraviroc is not recommended to be used in treatment-naïve patients based on the results of a clinical study in this population (see section 5.1).
Dose adjustment
Physicians should ensure that appropriate dose adjustment of maraviroc is made when maraviroc is co-administered with potent CYP3A4 inhibitors and/or inducers since maraviroc concentrations and its therapeutic effects may be affected (see sections 4.2 and 4.5). Please also refer to the respective Summary of Product Characteristics of the other antiretroviral medicinal products used in the combination.
Osteonecrosis
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Potential effect on immunity
CCR5 antagonists could potentially impair the immune response to certain infections. This should be taken into consideration when treating infections such as active tuberculosis and invasive fungal infections. The incidence of AIDS-defining infections was similar between maraviroc and placebo arms in the pivotal studies.
Soya lecithin
CELSENTRI contains soya lecithin.
If a patient is hypersensitive to peanut or soya, CELSENTRI should not be used.
Interaction with other medicinal products and other forms of interaction
Maraviroc is metabolised by cytochrome P450 CYP3A4 and CYP3A5. Co-administration of maraviroc with medicinal products that induce CYP3A4 may decrease maraviroc concentrations and reduce its therapeutic effects. Co-administration of maraviroc with medicinal products that inhibit CYP3A4 may increase maraviroc plasma concentrations. Dose adjustment of maraviroc is recommended when maraviroc is co-administered with potent CYP3A4 inhibitors and/or inducers. Further details for concomitantly administered medicinal products are provided below (see Table 2).
Maraviroc is a substrate for the transporters P-glycoprotein and OATP1B1, but the effect of these transporters on the exposure to maraviroc is not known.
Based on the in vitro and clinical data, the potential for maraviroc to affect the pharmacokinetics of co-administered medicinal products is low. In vitro studies have shown that maraviroc does not inhibit OATP1B1, MRP2 or any of the major P450 enzymes at clinically relevant concentrations (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4). Maraviroc had no clinically relevant effect on the pharmacokinetics of midazolam, the oral contraceptives ethinylestradiol and levonorgestrel, or urinary 6β-hydroxycortisol/cortisol ratio, suggesting no inhibition or induction of CYP3A4 in vivo. At higher exposure of maraviroc a potential inhibition of CYP2D6 cannot be excluded.
Renal clearance accounts for approximately 23% of total clearance of maraviroc when maraviroc is administered without CYP3A4 inhibitors. In vitro studies have shown that maraviroc does not inhibit any of the major renal uptake transporters at clinically relevant concentrations (OAT1, OAT3, OCT2, OCTN1, and OCTN2). Additionally, co-administration of maraviroc with tenofovir (substrate for renal elimination) and cotrimoxazole (contains trimethoprim, a renal cation transport inhibitor), showed no effect on the pharmacokinetics of maraviroc. In addition, co-administration of maraviroc with lamivudine/zidovudine showed no effect of maraviroc on lamivudine (primarily renally cleared) or zidovudine (non-P450 metabolism and renal clearance) pharmacokinetics. Maraviroc inhibits P-glycoprotein in vitro (IC50 is 183 μM). However, maraviroc does not significantly affect the pharmacokinetics of digoxin in vivo. It may not be excluded that maraviroc can increase the exposure to the P-glycoprotein substrate dabigatran etexilate.
Table 2. Interactions and adulta dose recommendations with other medicinal products:





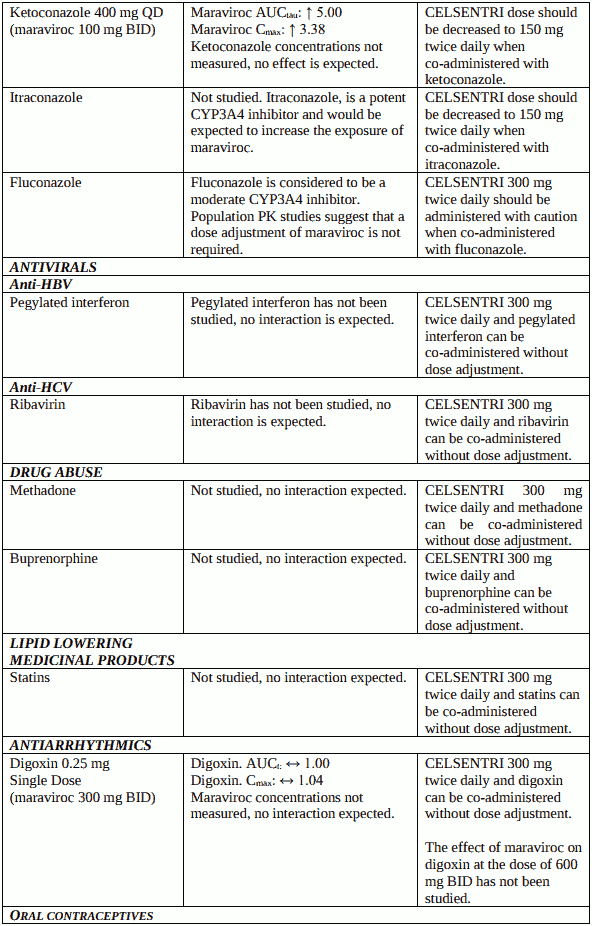

Fertility, pregnancy and lactation
Pregnancy
There are limited data from the use of maraviroc in pregnant women. The effect of maraviroc on human pregnancy is unknown. Studies in animals showed reproductive toxicity at high exposures. Primary pharmacological activity (CCR5 receptor affinity) was limited in the species studied (see section 5.3). Maraviroc should be used during pregnancy only if the expected benefit justifies the potential risk to the foetus.
Breast-feeding
It is unknown whether maraviroc is excreted in human milk. Available toxicological data in animals has shown extensive excretion of maraviroc in milk. Primary pharmacological activity (CCR5 receptor affinity) was limited in the species studied (see section 5.3). A risk to the newborn/infants cannot be excluded.
It is recommended that mothers infected by HIV do not breast-feed their infants under any circumstances in order to avoid transmission of HIV.
Fertility
There is no data on the effects of maraviroc on human fertility. In rats, there were no adverse effects on male or female fertility (see section 5.3).
Effects on ability to drive and use machines
Maraviroc may have a minor influence on the ability to drive and use machines. Patients should be informed that dizziness has been reported during treatment with maraviroc. The clinical status of the patient and the adverse reaction profile of maraviroc should be borne in mind when considering the patient’s ability to drive, cycle or operate machinery.
Undesirable effects
Summary of the safety profile
Adults
Assessment of treatment related adverse reactions is based on pooled data from two Phase 2b/3 studies in treatment-experienced adult patients (MOTIVATE 1 and MOTIVATE 2) and one study in treatment-naïve adult patients (MERIT) infected with CCR5-tropic HIV-1 (see sections 4.4 and 5.1).
The most frequently reported adverse reactions occurring in the Phase 2b/3 studies were nausea, diarrhoea, fatigue and headache. These adverse reactions were common (≥1/100 to <1/10).
Tabulated list of adverse reactions
The adverse reactions are listed by system organ class (SOC) and frequency. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1000 to <1/100), rare (≥1/10,000 to <1/1,000), not known (cannot be estimated from the available data). The adverse reactions and laboratory abnormalities presented below are not exposure adjusted.
Table 3. Adverse reactions observed in clinical trials or post-marketing:
Infections and infestations
Uncommon: Pneumonia, oesophageal candidiasis
Neoplasm benign, malignant and unspecified (including cysts and polyps)
Rare: Bile duct cancer, diffuse large B-cell lymphoma, Hodgkin’s disease, metastases to bone, metastases to liver, metastases to peritoneum, nasopharyngeal cancer, oesophageal carcinoma
Blood and lymphatic system disorders
Common: Anaemia
Rare: Pancytopenia, granulocytopenia
Metabolism and nutrition disorders
Common: Anorexia
Psychiatric disorders
Common: Depression, insomnia
Nervous system disorders
Uncommon: Seizures and seizure disorders
Cardiac disorders
Rare: Angina pectoris
Vascular disorders
Uncommon: Postural hypotension (see section 4.4)
Gastrointestinal disorders
Common: Abdominal pain, flatulence, nausea
Hepatobiliary disorders
Common: Alanine aminotransferase increased, aspartate aminotransferase increased
Uncommon: Hyperbilirubinaemia, gammaglutamyltransferase increased
Rare: Hepatitis toxic, hepatic failure, hepatic cirrhosis, blood alkaline phosphatase increased
Very rare: Hepatic failure with allergic features
Skin and subcutaneous tissue disorders
Common: Rash
Rare/not known: Stevens-Johnson syndrome/Toxic epidermal necrolysis
Musculoskeletal and connective tissue disorders
Uncommon: Myositis, blood creatine phosphokinase increased
Rare: Muscle atrophy
Renal and urinary disorders
Uncommon: Renal failure, proteinuria
General disorders and administration site conditions
Common: Asthenia
General disorders and administration site conditions
Common: Asthenia
Description of selected adverse reactions
Delayed type hypersensitivity reactions, typically occurring within 2-6 weeks after start of therapy and including rash, fever, eosinophilia and liver reactions have been reported (see also section 4.4). Skin and liver reactions can occur as single events, or in combination.
In HIV infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
Cases of syncope caused by postural hypotension have been reported.
Laboratory abnormalities
Table 4 shows the incidence ≥1% of Grade 3-4 Abnormalities (ACTG Criteria) based on the maximum shift in laboratory test values without regard to baseline values.
Table 4. Incidence ≥1% of grade 3-4 abnormalities (ACTG criteria) based on maximum shift in laboratory test values without regard to baseline studies MOTIVATE 1 and MOTIVATE 2 (pooled analysis, up to 48 weeks):
| Laboratory parameter | Limit | Maraviroc 300 mg twice daily + OBT N=421* (%) | Placebo + OBT N=207* (%) |
|---|---|---|---|
| Hepatobiliary disorders | |||
| Aspartate aminotransferase | >5.0x ULN | 4.8 | 2.9 |
| Alanine aminotransferase | >5.0x ULN | 2.6 | 3.4 |
| Total bilirubin | >5.0x ULN | 5.5 | 5.3 |
| Gastrointestinal disorders | |||
| Amylase | >2.0x ULN | 5.7 | 5.8 |
| Lipase | >2.0x ULN | 4.9 | 6.3 |
| Blood and lymphatic system disorders | |||
| Absolute neutrophil count | <750/mm3 | 4.3 | 1.9 |
ULN: Upper Limit of Normal
OBT: Optimised Background Therapy
* Percentages based on total patients evaluated for each laboratory parameter
The MOTIVATE studies were extended beyond 96 weeks, with an observational phase extended to 5 years in order to assess the long term safety of maraviroc. The Long Term Safety/Selected Endpoints (LTS/SE) included death, AIDS-defining events, hepatic failure, Myocardial infarction/cardiac ischaemia, malignancies, rhabdomyolysis and other serious infectious events with maraviroc treatment. The incidence of these selected endpoints for subjects on maraviroc in this observational phase was consistent with the incidence seen at earlier timepoints in the studies.
In treatment-naïve patients, the incidence of grade 3 and 4 laboratory abnormalities using ACTG criteria was similar among the maraviroc and efavirenz treatment groups.
Paediatric population
The adverse reaction profile in paediatric patients is based on 48 Week safety data from study A4001031 in which 103 HIV-1 infected, treatment-experienced patients aged 2 to <18 years received maraviroc twice-daily with optimised background therapy (OBT). Overall, the safety profile in paediatric patients was similar to that observed in adult clinical studies.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.