CONSION XL Prolonged-release capsule, hard Ref.[8035] Active ingredients: Galantamine
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2018 Publisher: Pharmathen S.A., Dervenakion 6, Pallini, Attiki, 153 51, Greece
Pharmacodynamic properties
Pharmacotherapeutic group: Antidementia drugs
ATC code: N06DA04
Mechanism of action
Galantamine, a tertiary alkaloid is a selective, competitive and reversible inhibitor of acetylcholinesterase. In addition, galantamine enhances the intrinsic action of acetylcholine on nicotinic receptors, probably through binding to an allosteric site of the receptor. As a consequence, an increased activity in the cholinergic system associated with improved cognitive function can be achieved in patients with dementia of the Alzheimer type.
Clinical studies
Galantamine was originally developed in the form of immediate-release tablets for twice-daily administration. The effective doses of galantamine in these placebo- controlled clinical trials with a duration of 5 to 6 months were 16, 24 and 32 mg/day. Of these doses 16 and 24 mg/day were determined to have the best benefit/risk relationship and are the recommended maintenance doses. The efficacy of galantamine has been shown using outcome measures which evaluate the three major symptom complexes of the disease and a global scale: the ADAS-cog/11 (a performance based measure of cognition), DAD and ADCS-ADL-Inventory (measurements of basic and instrumental Activities of Daily Living), the Neuropsychiatric Inventory (a scale that measures behavioural disturbances) and the CIBIC-plus (a global assessment by an independent physician based on a clinical interview with the patient and caregiver).
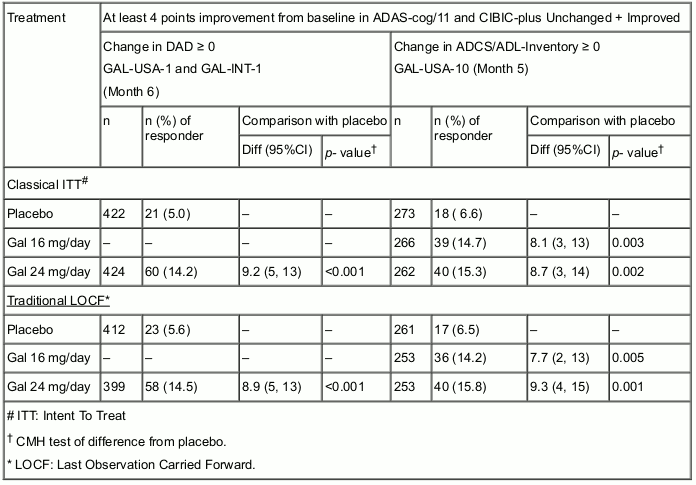
Composite Responder Analysis Based on at Least 4 Points Improvement in ADAS-cog/11 Compared to Baseline and CIBIC-plus Unchanged + Improved (1-4), and DAD/ADL Score Unchanged + Improved. See Table below:
The efficacy of galantamine prolonged-release capsules was studied in a randomised, double-blind, placebo-controlled trial, GAL-INT-10, using a 4-week dose escalation, flexible dosing regimen of 16 or 24 mg/day for a treatment duration of 6 months.
Galantamine immediate-release tablets (Gal-IR) were added as a positive control arm. Efficacy was evaluated using the ADAS-cog/11 and the CIBIC-plus scores as co- primary efficacy criteria, and ADCS-ADL and NPI scores as secondary end-points.
Galantamine prolonged-release capsules (Gal-PR) demonstrated statistically significant improvements in the ADAS-cog/11 score compared to placebo, but were not statistically different in the CIBIC-plus score compared to placebo. The results of the ADCS-ADL score were statistically significantly better compared to placebo at week 26.
Composite Responder Analysis at Week 26 Based on at Least 4 Points Improvement from Baseline in ADAS-cog/11, Total ADL Score Unchanged + Improved (≥0) and No Worsening in CIBIC-plus Score (1-4). See Table below:
| GAL-INT-10 | Placebo | Gal-IR† | Gal-PR* | p-value (Gal-PR* vs. Placebo) |
|---|---|---|---|---|
| (n=245) | (n=225) | (n=238) | ||
| Composite Response: n (%) | 20 (8.2) | 43 (19.1) | 38 (16.0) | 0.008 |
† Immediate-release tablets
* Prolonged-release capsules
Vascular dementia or Alzheimer’s disease with cerebrovascular disease
The results of a 26-week double-blind placebo-controlled trial, in which patients with vascular dementia and patients with Alzheimer’s disease and concomitant cerebrovascular disease (“mixed dementia”) were included, indicate that the symptomatic effect of galantamine is maintained in patients with Alzheimer’s disease and concomitant cerebrovascular disease (see section 4.4). In a post-hoc subgroup analysis, no statistically significant effect was observed in the subgroup of patients with vascular dementia alone.
In a second 26-week placebo-controlled trial in patients with probable vascular dementia, no clinical benefit of galantamine treatment was demonstrated.
Pharmacokinetic properties
Galantamine is an alkalinic compound with one ionisation constant (pKa 8.2). It is slightly lipophilic and has a partition coefficient (Log P) between n-octanol/buffer solution (pH 12) of 1.09. The solubility in water (pH 6) is 31 mg/mL. Galantamine has three chiral centres. The S, R, S-form is the naturally occurring form. Galantamine is partially metabolised by various cytochromes, mainly CYP2D6 and CYP3A4.
Some of the metabolites formed during the degradation of galantamine have been shown to be active in vitro but are of no importance in vivo.
Absorption
The absolute bioavailability of galantamine is high, 88.5 ± 5.4%. Galantamine prolonged-release capsules are bioequivalent to the twice-daily immediate-release tablets with respect to AUC24h and Cmin. The Cmax value is reached after 4.4 hours and is about 24% lower than that of the tablet. Food has no significant effect on AUC of the prolonged-release capsules. Cmax was increased by about 12% and Tmax increased by about 30 minutes when the capsule was given after food. However, these changes are unlikely to be clinically significant.
Distribution
The mean volume of distribution is 175 L. Plasma protein binding is low, 18%.
Biotransformation
Up to 75% of galantamine dosed is eliminated via metabolism. In vitro studies indicate that CYP2D6 is involved in the formation of O-desmethylgalantamine and CYP3A4 is involved in the formation of N-oxide-galantamine. The levels of excretion of total radioactivity in urine and faeces were not different between poor and extensive CYP2D6 metabolisers. In plasma from poor and extensive metabolisers, unchanged galantamine and its glucuronide accounted for most of the sample radioactivity. None of the active metabolites of galantamine (norgalantamine, O- desmethylgalantamine and O-desmethyl-norgalantamine) could be detected in their unconjugated form in plasma from poor and extensive metabolisers after single dosing. Norgalantamine was detectable in plasma from patients after multiple dosing, but did not represent more than 10% of the galantamine levels. In vitro studies indicated that the inhibition potential of galantamine with respect to the major forms of human cytochrome P450 is very low.
Elimination
Galantamine plasma concentration declines bi-exponentially, with a terminal half-life around 8-10 hours in healthy subjects. Typical oral clearance in the target population is about 200 mL/min with intersubject variability of 30% as derived from the population analysis of immediate-release tablets. Seven days after a single oral dose of 4 mg 3H-galantamine, 90-97% of the radioactivity is recovered in urine and 2.2-6.3% in faeces. After i.v. infusion and oral administration, 18-22% of the dose was excreted as unchanged galantamine in the urine in 24 hours, with a renal clearance of 68.4 ± 22.0 mL/min, which represents 20-25% of the total plasma clearance.
Dose-Linearity
Galantamine pharmacokinetics of galantamine prolonged-release capsules are dose proportional within the studied dose range of 8 mg to 24 mg once-daily in elderly and young age groups.
Characteristics in patients with Alzheimer’s disease
Data from clinical trials in patients indicate that the plasma concentrations of galantamine in patients with Alzheimer’s disease are 30% to 40% higher than in healthy young subjects primarily due to the advanced age and reduced kidney function. Based upon the population pharmacokinetic analysis, clearance in female subjects is 20% lower as compared to males. The galantamine clearance in poor metabolisers of CYP2D6 is about 25% lower than in extensive metabolisers, but no bimodality in the population is observed. Therefore, the metabolic status of the patient is not considered to be of clinical relevance in the overall population.
Special populations
Renal impairment
Elimination of galantamine decreases with decreasing creatinine clearance as observed in a study with renally impaired subjects. Compared to Alzheimer patients, peak and trough plasma concentrations are not increased in patients with a creatinine clearance of ≥9 mL/min. Therefore, no increase in adverse events is expected and no dose adjustments are needed (see section 4.2).
Hepatic impairment
The pharmacokinetics of galantamine in subjects with mild hepatic impairment (Child-Pugh score of 5-6) were comparable to those in healthy subjects. In patients with moderate hepatic impairment (Child-Pugh score of 7-9), AUC and half-life of galantamine were increased by about 30% (see section 4.2).
Pharmacokinetic/pharmacodynamic relationship
No apparent correlation between average plasma concentrations and efficacy parameters (i.e. change in ADAS-cog/11 and CIBIC-plus at month 6) were observed in the large Phase III trials with a dose-regimen of 12 and 16 mg twice-daily.
Plasma concentrations in patients experiencing syncope were within the same range as in the other patients at the same dose.
The occurrence of nausea is shown to correlate with higher peak plasma concentrations (see section 4.5).
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenic potential.
Reproduction toxicity studies showed a slight delay in development in rats and rabbits, at doses that are below the threshold of toxicity in the pregnant females.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.