DUPIXENT 300 mg Solution for injection Ref.[8955] Active ingredients: Dupilumab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Sanofi Winthrop Industrie, 82 avenue Raspail, 94250 Gentilly, France
Pharmacodynamic properties
Pharmacotherapeutic group: Other dermatological preparations, agents for dermatitis, excluding corticosteroids
ATC code: D11AH05
Mechanism of action
Dupilumab is a recombinant human IgG4 monoclonal antibody that inhibits interleukin-4 and interleukin-13 signalling. Dupilumab inhibits IL-4 signaling via the Type I receptor (IL-4Rα/γc), and both IL-4 and IL-13 signaling through the Type II receptor (IL-4Rα/IL-13Rα). IL-4 and IL-13 are major drivers of human type 2 inflammatory disease, such as atopic dermatitis, asthma, CRSwNP, PN, EoE, COPD, and CSU. Blocking the IL-4/IL-13 pathway with dupilumab in patients decreases many of the mediators of type 2 inflammation.
Pharmacodynamic effects
In atopic dermatitis clinical trials, treatment with dupilumab was associated with decreases from baseline in concentrations of type 2 immunity biomarkers, such as thymus and activation-regulated chemokine (TARC/CCL17), total serum IgE and allergen-specific IgE in serum. A reduction of lactate dehydrogenase (LDH), a biomarker associated with AD disease activity and severity, was observed with dupilumab treatment in adults and adolescents with atopic dermatitis.
In adult and adolescent patients with asthma, dupilumab treatment relative to placebo markedly decreased FeNO and circulating concentrations of eotaxin-3, total IgE, allergen specific IgE, TARC, and periostin, the type 2 biomarkers evaluated in clinical trials. These reductions in type 2 inflammatory biomarkers were comparable for the 200 mg Q2W and 300 mg Q2W regimens. In paediatric (6 to 11 years of age) patients with asthma, dupilumab treatment relative to placebo markedly decreased FeNO and circulating concentrations of total IgE, allergen specific IgE, and TARC, the type 2 biomarkers evaluated in clinical trials. These markers were near maximal suppression after 2 weeks of treatment, except for IgE which declined more slowly. These effects were sustained throughout treatment.
In COPD patients, dupilumab treatment decreased type 2 biomarkers including FeNO and total IgE compared to placebo. Decreases in FeNO were observed by Week 4. These effects on type 2 biomarkers were sustained throughout treatment with dupilumab.
Clinical efficacy and safety in atopic dermatitis
Adults with atopic dermatitis
The efficacy and safety of dupilumab as monotherapy and with concomitant topical corticosteroids were evaluated in three pivotal randomised, double-blind, placebo-controlled studies (SOLO 1, SOLO 2, and CHRONOS) in 2,119 patients 18 years of age and older with moderate to severe atopic dermatitis (AD) defined by Investigator's Global Assessment (IGA) score ≥3, an Eczema Area and Severity Index (EASI) score ≥16, and a minimum body surface area (BSA) involvement of ≥10%. Eligible patients enrolled into the three studies had previous inadequate response to topical medication.
In all three studies, patients received dupilumab subcutaneous (SC) injections administered as 1) an initial dose of 600 mg dupilumab (two 300 mg injections) on day 1, followed by 300 mg once every two weeks (Q2W); or 2) an initial dose of 600 mg dupilumab on day 1, followed by 300 mg once weekly (QW); or 3) matching placebo. If needed to control intolerable symptoms of atopic dermatitis, patients were permitted to receive rescue treatment (which included higher potency topical steroids or systemic immunosuppressants) at the discretion of the investigator. Patients who received rescue treatment were considered non-responders.
Endpoints
In all three pivotal studies, the co-primary endpoints were the proportion of patients with IGA 0 or 1 ("clear" or "almost clear") with a reduction of ≥2 points on a 0-4 IGA scale and the proportion of patients with improvement of at least 75% in EASI (EASI-75). Key secondary and other clinically relevant secondary endpoints are presented in Table 8.
Baseline Characteristics
In the monotherapy studies (SOLO 1 and SOLO 2), across all treatment groups, the mean age was 38.3, the mean weight was 76.9 kg, 42.1% were female, 68.1% were white, 21.8% were Asian, and 6.8% were black. In these studies, 51.6% of patients had a baseline IGA score of 3 (moderate AD), 48.3% of patients had a baseline IGA of 4 (severe AD) and 32.4% of patients had received prior systemic immunosuppressants. The baseline mean EASI score was 33.0, the baseline weekly averaged pruritus Numerical Rating Scale (NRS) was 7.4, the baseline mean POEM score was 20.5, the baseline mean DLQI was 15.0, and the baseline mean HADS total score was 13.3.
In the concomitant TCS study (CHRONOS), across all treatment groups, the mean age was 37.1, the mean weight was 74.5 kg, 39.7% were female, 66.2% were white, 27.2% were Asian, and 4.6% were black. In this study, 53.1% of patients had a baseline IGA score of 3 and 46.9 % of patients had a baseline IGA of 4 and 33.6% of patients received prior systemic immunosuppressants. The baseline mean EASI score was 32.5, the baseline weekly pruritus NRS was 7.3, the baseline mean POEM score was 20.1, the baseline mean DLQI was 14.5, and the baseline mean HADS total score was 12.7.
Clinical Response
16-week monotherapy studies (SOLO 1 and SOLO 2) and 52-week concomitant TCS study (CHRONOS)
In SOLO 1, SOLO 2, and CHRONOS from baseline to week 16, a significantly greater proportion of patients randomised to dupilumab achieved an IGA 0 or 1 response, EASI-75, and/or an improvement of >4 points on the pruritus NRS (key secondary endpoint) compared to placebo (see Table 8).
A significantly greater proportion of patients randomised to dupilumab alone or with TCS achieved a rapid improvement in the pruritus NRS compared to placebo or placebo + TCS (defined as ≥4-point improvement as early as week 2, p<0.01 and p<0.05, respectively).
A persistent treatment effect of dupilumab was observed in the CHRONOS study up to week 52 (see Table 8).
The efficacy results for co-primary, key secondary and other clinically relevant secondary endpoints for all three studies are presented in Table 8.
Table 8. Efficacy results of dupilumab monotherapy at week 16 (FAS) and with concomitant TCSa at week 16 and week 52:
| SOLO 1 Week 16 (FAS)b | SOLO 2 Week 16 (FAS)b | CHRONOS Week 16 (FAS)h | CHRONOS Week 52 (FAS Week 52)h | |||||
|---|---|---|---|---|---|---|---|---|
| Placebo | Dupilumab 300 mg Q2W | Placebo | Dupilumab 300 mg Q2W | Placebo + TCS | Dupilumab 300 mg Q2 W + TCS | Placebo + TCS | Dupilumab 300 mg Q2W + TCS | |
| Patients randomised | 224 | 224 | 236 | 233 | 315 | 106 | 264 | 89 |
| IGA 0 or 1c, % respondersd | 10.3% | 37.9%g | 8.5% | 36.1%g | 12.4% | 38.7%g | 12.5% | 36.0%g |
| EASI-50. % respondersd | 24.6% | 68.8%g | 22.0% | 65.2%g | 37.5% | 80.2%j | 29.9% | 78.7%j |
| EASI-75. % respondersd | 14.7% | 51.3%g | 11.9% | 44.2%g | 23.2% | 68.9%g | 21.6% | 65.2%g |
| EASI-90. % respondersd | 7.6% | 35.7%g | 7.2% | 30.0%g | 11.1% | 39.6%j | 15.5% | 50.6%j |
| Pruritus NRS, LS mean% change from baseline (+/- SE) | -26.1% (3.02) | -51.0%g (2.50) | -15.4% (2.98) | -44.3%g (2.28) | -30.3% (2.36) | -56.6%g (3.95) | -31.7% (3.95) | -57.0%i (6.17) |
| Pruritus NRS (≥4-point improvement),% respondersd,e,f | 12.3% (26/212) | 40.8%g (87/213) | 9.5% (21/221) | 36.0%g (81/225) | 19.7% (59/299) | 58.8%g (60/102) | 12.9% (32/249) | 51.2%g (44/86) |
LS = least squares; SE= standard error
a all patients were on background topical corticosteroids therapy and patients were permitted to use topical calcineurin inhibitors.
b full analysis set (FAS) includes all patients randomised.
c responder was defined as a patient with IGA 0 or 1 ("clear" or "almost clear") with a reduction of >2 points on a 0-4 IGA scale.
d patients who received rescue treatment or with missing data were considered as non-responders.
e the number of patients with baseline pruritus NRS ≥4 as denominator.
f a significantly greater proportion of patients on dupilumab had improvement in pruritus NRS of ≥4 points compared to placebo at week 2 (p<0.01).
g p-value <0.0001. statistically significant vs placebo with adjustment for multiplicity.
h full analysis set (FAS) includes all patients randomised. FAS week 52 includes all patients randomised at least one year before the cutoff date of the primary analysis.
i nominal p-value = 0.0005
j nominal p-value <0.0001
In SOLO1, SOLO2 and CHRONOS similar results were observed in patients receiving Dupilumab 300 mg QW.
Figure 1a and Figure 1b show the mean percent change from baseline in EASI and the mean percent change from baseline in NRS respectively up to week 16 in SOLO1 and SOLO2.
Figure 2a and Figure 2b show the mean percent change from baseline in EASI and the mean percent change from baseline in NRS, respectively up to week 52 in CHRONOS.
Figure 1. Mean percent change from baseline in EASI in SOLO 16a and SOLO 2a (FAS)b:

LS = least squares
a In the primary analyses of the efficacy endpoints, patients who received rescue treatment or with missing data were considered non-responders.
b Full analysis set (FAS) includes all patients randomised.
Figure 2. Mean percent change from baseline in NRS in SOLO 1a and SOLO 2a (FAS)b:

LS = least squares
a In the primary analyses of the efficacy endpoints, patients who received rescue treatment or with missing data were considered non-responders.
b FAS week 52 includes all patients randomised at least one year before the cutoff date of the primary analysis.
Treatment effects in subgroups (weight, age, gender, race, and background treatment, including immunosuppressants) in SOLO 1, SOLO 2, and CHRONOS were consistent with the results in the overall study population within each of these studies.
Clinical response in patients not adequately controlled with, intolerant to, or for whom ciclosporin treatment was inadvisable (CAFE study)
CAFE study evaluated the efficacy of dupilumab compared to placebo during a 16-week treatment period, administered with concomitant TCS, in adult patients with AD who are not adequately controlled with, or are intolerant to, oral ciclosporin, or when this treatment is currently contraindicated or not medically advisable.
A total of 325 patients were enrolled, with 210 patients who were previously exposed to ciclosporin and 115 patients who have never been exposed to ciclosporin because ciclosporin treatment was medically inadvisable. The mean age was 38.4 years, 38.8% were female, the baseline mean EASI score was 33.1, the mean BSA was 55.7, the baseline weekly average pruritis NRS was 6.4, and the baseline mean DLQI was 13.8.
Primary endpoint (proportion of patients with EASI-75) and secondary endpoints for the 16 week CAFE study are summarized in Table 9.
Table 9. Results of the primary and secondary endpoints in CAFE study:
| Placebo + TCS | Dupilumab 300 mg Q2W + TCS | Dupilumab 300 mg QW+TCS | |
|---|---|---|---|
| Patients randomised | 108 | 107 | 110 |
| EASI-75.% responders | 29.6% | 62.6% | 59.1% |
| EASI, LS mean % change from baseline (+/- SE) | -46.6 (2.76) | -79.8 (2.59) | -78.2 (2.55) |
| Pruritus NRS, LS mean % change from baseline (+/- SE) | -25.4% (3.39) | -53.9% (3.14) | -51.7% (3.09) |
| DLQI, LS mean change from baseline (SE) | -4.5 (0.49) | -9.5 (0.46) | -8.8 (0.45) |
(all p-values <0.0001. statistically significant vs placebo with adjustment for multiplicity.)
In the subgroup of patients resembling the CAFE study population within the 52 week CHRONOS study, 69.6% of dupilumab 300 mg Q2W-treated patients reached EASI-75 vs 18.0% placebo-treated patients at week 16. and 52.4% of dupilumab 300 mg Q2W-treated vs 18.6% placebo-treated at week 52. In this subset, the percent change of pruritus NRS from baseline was -51.4% vs -30.2% at week 16 and -54.8% vs -30.9% at week 52. for the dupilumab 300 mg Q2W and placebo groups respectively.
Maintenance and durability of response (SOLO CONTINUE study)
To evaluate maintenance and durability of response, subjects treated with dupilumab for 16 weeks in SOLO 1 and SOLO 2 studies who achieved IGA 0 or 1 or EASI-75 were re-randomised in SOLO CONTINUE study to an additional 36-week treatment of dupilumab or placebo, for a cumulative 52- week study treatment. Endpoints were assessed at weeks 51 or 52.
The co-primary endpoints were the difference between baseline (week 0) and week 36 in percent change in EASI from SOLO 1 and SOLO 2 studies baseline and percentage of patients with EASI-75 at week 36 in patients with EASI-75 at baseline.
Patients who continued on the same dose regimen received in the SOLO 1 and SOLO 2 studies (300 mg Q2W or 300 mg QW) showed the optimal effect in maintaining clinical response while efficacy for other dose regimens diminished in a dose-dependent manner.
Primary and secondary endpoints for the 52 week SOLO CONTINUE study are summarized in Table 10.
Table 10. Results of the primary and secondary endpoints in SOLO CONTINUE study:
| Placebo | Dupilumab 300 mg | |||
|---|---|---|---|---|
| N=83 | Q8W N=84 | Q4W N=86 | Q2W/QW N=169 | |
| Co-Primary Endpoints | ||||
| LS mean change (SE) between baseline and week 36 in percent change in EASI Score from Parent Study baseline | 21.7 (3.13) | 6.8*** (2.43) | 3.8*** (2.28) | 0.1*** (1.74) |
| Percent of patients with EASI-75 at week 36 for patients with EASI-75 at baseline, n (%) | 24/79 (30.4%) | 45/82* (54.9%) | 49/84** (58.3%) | 116/162*** (71.6%) |
| Key Secondary Endpoints | ||||
| Percent of patients whose IGA response at week 36 was maintained within 1 point of baseline in the subset of patients with IGA (0.1) at baseline, n (%) | 18/63 (28.6) | 32/64† (50.0) | 41/66** (62.1) | 89/126*** (70.6) |
| Percent of patients with IGA (0.1) at week 36 in the subset of patients with IGA (0.1) at baseline, n (%) | 9/63 (14.3) | 21/64† (32.8) | 29/66** (43.9) | 68/126*** (54.0) |
| Percent of patients whose peak pruritus NRS increased by ≥3 points from baseline to week 35 in the subset of patients with peak pruritus NRS ≤7 at baseline, n (%) | 56/80 (70.0) | 45/81 (55.6) | 41/83† (49.4) | 57/168*** (33.9) |
† p-value <0.05.
* p-value <0.01.
** p-value <0.001.
*** p-value ≤0.0001 (all statistically significant vs placebo with adjustment for multiplicity.)
In SOLO CONTINUE, a trend for increased treatment-emergent ADA positivity with increased dosing intervals was observed. Treatment-emergent ADA: QW: 1.2%; Q2W: 4.3%; Q4W: 6.0%; Q8W: 11.7%. ADA responses lasting more than 12 weeks: QW: 0.0%; Q2W: 1.4%; Q4W: 0.0%; Q8W: 2.6%.
Quality of life/patient-reported outcomes in atopic dermatitis
In both monotherapy studies (SOLO 1 and SOLO 2), both dupilumab 300 mg Q2W and 300 mg QW groups significantly improved patient-reported symptoms and the impact of AD on sleep, anxiety and depression symptoms as measured by HADS, and health-related quality of life as measured by POEM and DLQI total scores, respectively, at 16 weeks compared to placebo (see Table 11).
Similarly, in the concomitant TCS study (CHRONOS), dupilumab 300 mg Q2W + TCS and dupilumab 300 mg QW + TCS improved patient-reported symptoms and the impact of AD on sleep and health-related quality of life as measured by POEM and DLQI total scores, respectively, at 52 weeks compared to placebo + TCS (see Table 11).
Table 11. Additional secondary endpoint results of dupilumab monotherapy at week 16 and concomitant use of TCS at week 16 and week 52:
| SOLO 1 Week 16 (FAS) | SOLO 2 Week 16 (FAS) | CHRONOS Week 16 (FAS) | CHRONOS Week 52 (FAS Week 52) | |||||
|---|---|---|---|---|---|---|---|---|
| Placebo | Dupilumab 300 mg Q2W | Placebo | Dupilumab 300 mg Q2W | Placebo +TCS | Dupilumab 300 mg Q2W + TCS | Placebo +TCS | Dupilumab 300 mg Q2W + TCS | |
| Patients randomized | 224 | 224 | 236 | 233 | 315 | 106 | 264 | 89 |
| DLQI, LS mean change from baseline (SE) | -5.3 (0.50) | -9.3a (0.40) | -3.6 (0.50) | -9.3a (0.38) | -5.8 (0.34) | -10.0f (0.50) | -7.2 (0.40) | -11.4f (0.57) |
| POEM, LS mean change from baseline (SE) | -5.1 (0.67) | -11.6a (0.49) | -3.3 (0.55) | -10.2a (0.49) | -5.3 (0.41) | -12.7f (0.64) | -7.0 (0.57) | -14.2f (0.78) |
| HADS, LS mean change from baseline (SE) | -3.0 (0.65) | -5.2b (0.54) | -0.8 (0.44) | -5.1a (0.39) | -4.0 (0.37) | -4.9c (0.58) | -3.8 (0.47) | -5.5e (0.71) |
| DLQI (≥4-point improvement), % respondersd | 30.5% (65/213) | 64.1%f (134/209) | 27.6% (62/225) | 73.1%f (163/223) | 43.0% (129/300) | 74.3%f (231/311) | 30.3% (77/254) | 80.0%f (68/85) |
| POEM (≥4-point improvement), % respondersd | 26.9% (60/223) | 67.6%f (150/222) | 24.4% (57/234) | 71.7%f (167/233) | 36.9% (115/312) | 77.4%f (246/318) | 26.1% (68/261) | 76.4%f (68/89) |
| Patients achieving HADS-anxiety and HADS- depression score <8.%d | 12.4% (12/97) | 41.0%f (41/100) | 6.1% (7/115) | 39.5%f (51/129) | 26.4% (39/148) | 47.4%g (73/154) | 18.0% (24/133) | 43.4%g (23/53) |
LS = least squares; SE = standard error
a p-value <0.0001. bp-value <0.001. cp-value <0.05 (all statistically significant vs placebo with adjustment for multiplicity.
d the number of patients with baseline pruritus DLQI, POEM, and HADS as denominator.
e nominal p-value <0.05. fnominal p-value <0.0001. gnominal p-value <0.001
In SOLO1, SOLO2 and CHRONOS similar results were observed in patients receiving Dupilumab 300 mg QW.
Adolescents with atopic dermatitis (12 to 17 years of age)
The efficacy and safety of dupilumab monotherapy in adolescent patients was evaluated in a multicentre, randomised, double-blind, placebo-controlled study (AD-1526) in 251 adolescent patients 12 to 17 years of age with moderate-to-severe atopic dermatitis (AD) defined by Investigator's Global Assessment (IGA) score ≥3 in the overall assessment of AD lesions on a severity scale of 0 to 4. an Eczema Area and Severity Index (EASI) score ≥16 on a scale of 0 to 72. and a minimum body surface area (BSA) involvement of ≥10%. Eligible patients enrolled into this study had previous inadequate response to topical medication.
Patients received dupilumab was administered by subcutaneous (SC) injections either as: 1) an initial dose of 400 mg dupilumab (two 200 mg injections) on day 1, followed by 200 mg once every other week (Q2W) for patients with baseline weight of <60 kg or an initial dose of 600 mg dupilumab (two 300 mg injections) on day 1, followed by 300 mg Q2W for patients with baseline weight of ≥60 kg; or 2) an initial dose of 600 mg dupilumab (two 300 mg injections) on day 1, followed by 300 mg every 4 weeks (Q4W) regardless of baseline body weight; or 3) matching placebo. If needed to control intolerable symptoms, patients were permitted to receive rescue treatment at the discretion of the investigator. Patients who received rescue treatment were considered non-responders.
In this study, the mean age was 14.5 years, the median weight was 59.4 kg, 41.0% were female, 62.5% were White, 15.1% were Asian, and 12.0% were Black. At baseline 46.2% of patients had a baseline IGA score of 3 (moderate AD), 53.8% of patients had a baseline IGA of 4 (severe AD), the mean BSA involvement was 56.5%, and 42.4% of patients had received prior systemic immunosuppressants. Also at baseline the mean Eczema Area and Severity Index (EASI) score was 35.5, the baseline weekly averaged pruritus Numerical Rating Scale (NRS) was 7.6, the baseline mean Patient Oriented Eczema Measure (POEM) score was 21.0, and the baseline mean Children Dermatology Life Quality Index (CDLQI) was 13.6. Overall, 92.0% of patients had at least one co-morbid allergic condition; 65.6% had allergic rhinitis, 53.6% had asthma, and 60.8% had food allergies.
The co-primary endpoint was the proportion of patients with IGA 0 or 1 ("clear" or "almost clear") least a 2-point improvement and the proportion of patients with EASI-75 (improvement of at least 75% in EASI), from baseline to week 16.
Clinical Response
The efficacy results at week 16 for adolescent atopic dermatitis study are presented in Table 12.
Table 12. Efficacy results of dupilumab in the adolescent atopic dermatitis study at week 16 (FAS):
| AD-1526 (FAS)a | ||
|---|---|---|
| Placebo | Dupilumab 200 mg (<60 kg) and 300 mg (≥60 kg) Q2W | |
| Patients randomised | 85a | 82a |
| IGA 0 ή 1b, % respondersc | 2.4% | 24.4%d |
| EASI-50. % respondersc | 12.9% | 61.0%d |
| EASI-75. % respondersc | 8.2% | 41.5%d |
| EASI-90. % respondersc | 2.4% | 23.2%d |
| EASI, LS mean % change from baseline (+/-SE) | -23.6% (5.49) | -65.9%d (3.99) |
| Pruritus NRS, LS mean % change from baseline (+/- SE) | -19.0% (4.09) | -47.9 %d (3.43) |
| Pruritus NRS (≥4-point improvement), % respondersc | 4.8% | 36.6%d |
| CDLQI, LS mean change from baseline (+/-SE) | -5.1 (0.62) | -8.5d (0.50) |
| CDLQI, (≥6-point improvement), % responders | 19.7% | 60.6%e |
| POEM, LS mean change from baseline (+/- SE) | -3.8 (0.96) | -10.1d (0.76) |
| POEM, (≥6-point improvement), % responders | 9.5% | 63.4%e |
a full Analysis Set (FAS) includes all patients randomised.
b responder was defined as a subject with IGA 0 or 1 ("clear" or "almost clear") with a reduction of ≥2 points on a 0-4 IGA scale.
c patients who received rescue treatment or with missing data were considered as non-responders (58.8% and 20.% in the placebo and dupilumab arms, respectively).
d p–value <0.0001 (statistically significant vs placebo with adjustment for multiplicity)
e nominal p-value <0.0001
A larger percentage of patients randomised to placebo needed rescue treatment (topical corticosteroids, systemic corticosteroids, or systemic non-steroidal immunosuppressants) as compared to the dupilumab group (58.8% and 20.7%, respectively).
A significantly greater proportion of patients randomised to dupilumab achieved a rapid improvement in the pruritus NRS compared to placebo (defined as ≥4-point improvement as early as week 4; nominal p<0.001) and the proportion of patients responding on the pruritus NRS continued to increase through the treatment period.
The dupilumab group significantly improved patient-reported symptoms, the impact of AD on sleep and health-related quality of life as measured by POEM and CDLQI scores at 16 weeks compared to placebo.
The long-term efficacy of dupilumab in adolescent patients with moderate-to-severe AD who had participated in previous clinical trials of dupilumab was assessed in open-label extension study (AD-1434). Efficacy data from this study suggests that clinical benefit provided at week 16 was sustained through week 52.
Paediatrics (6 to 11 years of age)
The efficacy and safety of dupilumab in paediatric patients concomitantly with TCS was evaluated in a multicentre, randomised, double-blind, placebo-controlled study (AD-1652) in 367 subjects 6 to 11 years of age, with severe AD defined by an IGA score of 4 (scale of 0 to 4), an EASI score ≥21 (scale of 0 to 72), and a minimum BSA involvement of ≥15%. Eligible patients enrolled into this trial had previous inadequate response to topical medication. Enrollment was stratified by baseline weight (<30 kg; ≥30 kg).
Patients in the dupilumab Q2W + TCS group with baseline weight of <30 kg received an initial dose of 200 mg on Day 1, followed by 100 mg Q2W from week 2 to week 14, and patients with baseline weight of ≥30 kg received an initial dose of 400 mg on Day 1, followed by 200 mg Q2W from week 2 to week 14. Patients in the dupilumab Q4W + TCS group received an initial dose of 600 mg on Day 1, followed by 300 mg Q4W from week 4 to week 12, regardless of weight.
In this study, the mean age was 8.5 years, the median weight was 29.8 kg, 50.1% of patients were female, 69.2% were White, 16.9% were Black, and 7.6% were Asian. At baseline, the mean BSA involvement was 57.6%, and 16.9% had received prior systemic non-steroidal immunosuppressants. Also, at baseline the mean EASI score was 37.9, and the weekly average of daily worst itch score was 7.8 on a scale of 0-10, the baseline mean SCORAD score was 73.6, the baseline POEM score was 20.9, and the baseline mean CDLQI was 15.1. Overall, 91.7% of subjects had at least one co-morbid allergic condition; 64.4% had food allergies, 62.7% had other allergies, 60.2% had allergic rhinitis, and 46.7% had asthma.
The co-primary endpoint was the proportion of patients with IGA 0 or 1 ("clear" or "almost clear") at least a 2-point improvement and the proportion of patients with EASI-75 (improvement of at least 75% in EASI), from baseline to week 16.
Clinical Response
Table 13 presents the results by baseline weight strata for the approved dose regimens.
Table 13. Efficacy results of dupilumab with concomitant TCS in AD-1652 at week 16 (FAS)a:
| Dupilumab 300 mg Q4Wd + TCS | Placebo + TCS | Dupilumab 200 mg Q2We + TCS | Placebo + TCS | |
|---|---|---|---|---|
| (N=122) | (N=123) | (N=59) | (N=62) | |
| ≥15 kg | ≥15 kg | ≥30 kg | ≥30 kg | |
| IGA 0 ή 1b, % respondersc | 32.8%f | 11.4% | 39.0%h | 9.7% |
| EASI-50, % respondersc | 91.0%f | 43.1% | 86.4%g | 43.5% |
| EASI-75, % respondersc | 69.7%f | 26.8% | 74.6%g | 25.8% |
| EASI-90, % respondersc | 41.8%f | 7.3% | 35.6%h | 8.1% |
| EASI, LS mean % change from baseline (+/-SE) | -82.1%f (2.37) | -48.6% (2.46) | -80.4%g (3.61) | -48.3% (3.63) |
| Pruritus NRS, LS mean % change from baseline (+/- SE) | -54.6%f (2.89) | -25.9% (2.90) | -58.2%g (4.01) | -25.0% (3.95) |
| Pruritus NRS (≥4-point improvement), % respondersc | 50.8%f | 12.3% | 61.4%g | 12.9% |
| CDLQI, LS mean change from baseline (+/-SE) | -10.6f (0.47) | -6.4 (0.51) | -9.8g (0.63) | -5.6 (0.66) |
| CDLQI, (≥6-point improvement), % responders | 77.3%g | 38.8% | 80.8%g | 35.8% |
| POEM, LS mean change from baseline (+/- SE) | -13.6f (0.65) | -5.3 (0.69) | -13.6g (0.90) | -4.7 (0.91) |
| POEM, (≥6-point improvement), % responders | 81.7%g | 32.0% | 79.3%g | 31.1 % |
a full Analysis Set (FAS) includes all patients randomised.
b responder was defined as a patient with an IGA 0 or 1 ("clear" or "almost clear").
c patients who received rescue treatment or with missing data were considered as non-responders.
d at Day 1. patients received 600 mg of dupilumab (see section 5.2).
e at Day 1. patients received 400 mg (baseline weight ≥30 kg) of dupilumab.
f p-value < 0.0001 (statistically significant vs placebo with adjustment for multiplicity)
g nominal p-values <0.0001
h nominal p-value = 0.0002
A greater proportion of patients randomised to dupilumab + TCS achieved an improvement in the peak pruritus NRS compared to placebo + TCS (defined as ≥4-point improvement at week 4).
The dupilumab groups significantly improved patient-reported symptoms, the impact of AD on sleep and health-related quality of life as measured by POEM and CDLQI scores at 16 weeks compared to placebo.
The long-term efficacy and safety of dupilumab + TCS in paediatric patients with moderate to severe atopic dermatitis who had participated in the previous clinical trials of dupilumab + TCS was assessed in an open-label extension study (AD-1434). Efficacy data from this trial suggests that clinical benefit provided at week 16 was sustained through week 52. Some patients receiving dupilumab 300 mg Q4W + TCS showed further clinical benefit when escalated to dupilumab 200 mg Q2W + TCS. The safety profile of dupilumab in patients followed through week 52 was similar to the safety profile observed at week 16 in the AD-1526 and AD-1652 studies.
Paediatrics (6 Months to 5 years of age)
The efficacy and safety of dupilumab + TCS in paediatric patients was evaluated in a multicentre, randomised, double-blind, placebo-controlled study (AD-1539) in 162 patients 6 months to 5 years of age, with moderate-to-severe AD (ITT population) defined by an IGA score ≥3 (scale of 0 to 4), an EASI score ≥16 (scale of 0 to 72), and a minimum BSA involvement of ≥10. Of the 162 patients, 125 patients had severe AD defined by an IGA score of 4. Eligible patients enrolled into this study had previous inadequate response to topical medication. Enrollment was stratified by baseline weight (≥5 to <15 kg and ≥15 to <30 kg).
Patients in the dupilumab Q4W + TCS group with baseline weight of ≥5 to <15 kg received an initial dose of 200 mg on Day 1, followed by 200 mg Q4W from week 4 to week 12, and patients with baseline weight of ≥15 to <30 kg received an initial dose of 300 mg on Day 1, followed by 300 mg Q4W from week 4 to week 12. Patients were permitted to receive rescue treatment at the discretion of the investigator. Patients who received rescue treatment were considered non-responders.
In AD-1539, the mean age was 3.8 years, the median weight was 16.5 kg, 38.9% of patients were female, 68.5% were White, 18.5% were Black, and 6.2% were Asian. At baseline, the mean BSA involvement was 58.4%, and 15.5% had received prior systemic non-steroidal immunosuppressants. Also, at baseline the mean EASI score was 34.1, and the weekly average of daily worst itch score was 7.6 on a scale of 0-10. Overall, 81.4% of patients had at least one co-morbid allergic condition; 68.3% had food allergies, 52.8% had other allergies, 44.1% had allergic rhinitis, and 25.5% had asthma.
These baseline disease characteristics were comparable between moderate-to-severe and severe AD populations.
The co-primary endpoint was the proportion of patients with IGA 0 or 1 ("clear" or "almost clear", at least a 2-point improvement) and the proportion of patients with EASI-75 (improvement of at least 75% in EASI), from baseline to week 16. The primary endpoint was the proportion of patients with an IGA 0 (clear) or 1 (almost clear) at week 16
Clinical Response
The efficacy results at week 16 for AD-1539 are presented in Table 14.
Table 14. Efficacy results of dupilumab with concomitant TCS in AD-1539 at Week 16 (FAS)a:
| Dupilumab 200 mg (5 to <15kg) or 300 mg (15 to <30 kg) Q4Wd+ TCS (ITT population)(N=83)a | Placebo + TCS (ITT population) (N=79) | Dupilumab 200 mg (5 to <15 kg) or 300 mg (15 to <30 kg) Q4Wd+ TCS (severe AD population) (N=63) | Placebo + TCS (severe AD population) (N=62) | |
|---|---|---|---|---|
| IGA 0 or 1b,c | 27.7%e | 3.9% | 14.3%f | 1.7% |
| EASI-50, % respondersc | 68.7%e | 20.2% | 60.3%g | 19.2% |
| EASI-75c | 53.0%e | 10.7% | 46.0%g | 7.2% |
| EASI-90c | 25.3%e | 2.8% | 15.9%h | 0% |
| EASI, LS mean % change from baseline (+/-SE) | -70.0%e (4.85) | -19.6% (5.13) | -55.4%g (5.01) | -10.3% (5.16) |
| Worst scratch/itch NRS, LS mean % change from baseline (+/-SE)* | -49.4%e (5.03) | -2.2% (5.22) | -41.8g (5.35) | 0.5 (5.40) |
| Worst Scratch/Itch NRS (≥4-point improvement)c* | 48.1%e | 8.9% | 42.3%i | 8.8% |
| Patient's sleep quality NRS, LS mean change from baseline (+/- SE)* | 2.0e (0.25) | 0.3 (0.26) | 1.7g (0.25) | 0.2 (0.25) |
| Patient's sleep quality NRS, LS mean change from baseline (+/- SE)* | -3.9e (0.30) | -0.6 (0.30) | -3.4g (0.29) | -0.3 (0.29) |
| POEM, LS mean change from baseline (+/- SE)* | -12.9e (0.89) | -3.8 (0.92) | -10.6g (0.93) | -2.5 (0.95) |
a Full Analysis Set (FAS) includes all patients randomised.
b Responder was defined as a patient with an IGA 0 or 1 ("clear" or "almost clear").
c Patients who received rescue treatment (62% and 19% in the placebo and dupilumab arms, respectively) or with missing data were considered as non-responders.
d At Day 1. patients received 200 mg (5 to <15kg) or 300 mg (15 to <30 kg) of dupilumab.
e p–values <0.0001.
f nominal p-value <0.05.
g nominal p-value <0.0001.
h nominal p-value <0.005.
i nominal pvalue <0.001
* Caregiver reported outcome
A significantly greater proportion of patients randomised to dupilumab + TCS achieved a rapid improvement in the Worst Scratch/Itch NRS compared to placebo + TCS (defined as ≥4-point improvement as early as week 3. nominal p<0.005) and the proportion of patients responding on the Worst Scratch/Itch NRS continued to increase through the treatment period.
In this study, dupilumab significantly improved health-related quality of life as measured by the CDLQI (in 85 patients 4 to 5 years old) and IDQOL (in 77 patients 6 months to 3 years old). In the ITT population, greater LS mean changes in CDLQI and IDQOL scores from baseline to week 16 were observed in the dupilumab + TCS (-10.0 and -10.9) group compared to the placebo + TCS group (-2.5 and -2.0), respectively (p<0.0001). Similar improvements in both CDLQI and IDQOL were observed in the severe AD population.
The long-term efficacy and safety of dupilumab + TCS in paediatric patients with moderate to severe atopic dermatitis who had participated in the previous clinical trials of dupilumab + TCS were assessed in an open-label extension study (AD-1434). Efficacy data from this trial suggest that clinical benefit provided at week 16 was sustained through week 52. The safety profile of dupilumab in patients followed through week 52 was similar to the safety profile observed at week 16 in the AD-1539 study.
Atopic Hand and Foot Dermatitis (adults and adolescents)
The efficacy and safety of dupilumab was evaluated in a 16-week multicenter, randomized, double-blind, parallel-group, placebo-controlled trial (AD-1924) in 133 adult and paediatric patients 12 to 17 years of age with moderate-to-severe atopic hand and foot dermatitis, defined by an IGA (hand and foot) score ≥3 (scale of 0 to 4) and a hand and foot Peak Pruritus Numeric Rating Scale (NRS) score for maximum itch intensity ≥4 (scale of 0 to 10). Eligible patients had previous inadequate response or intolerance to treatment of hand and foot dermatitis with topical AD medications.
In AD-1924, 38% of patients were male, 80% were White, 72% of subjects had a baseline IGA (hand and foot) score of 3 (moderate atopic hand and foot dermatitis), and 28% of patients had a baseline IGA (hand and foot) score of 4 (severe atopic hand and foot dermatitis). The baseline weekly averaged hand and foot Peak Pruritus NRS score was 7.1.
The primary endpoint was the proportion of patients with an IGA hand and foot score of 0 (clear) or 1 (almost clear) at Week 16. The key secondary endpoint was reduction of itch as measured by the hand and foot Peak Pruritus NRS (≥4-point improvement). Other patient reported outcomes included assessment of hand and foot skin pain NRS (0-10), quality of sleep NRS (0-10), quality of life in Hand Eczema Questionnaire (0-117) (QoLHEQ) and work productivity and impairment (WPAI) (0-100%).
The proportion of patients with an IGA (hand and foot) 0 to 1 at Week 16 was 40.3% for dupilumab and 16.7% for placebo (treatment difference 23.6, 95% CI: 8.84, 38.42). The proportion of patients with improvement (reduction) of weekly averaged hand and foot Peak Pruritus NRS ≥4 at Week 16 was 52.2% for dupilumab and 13.6% for placebo (treatment difference 38.6, 95% CI: 24.06, 53.15). Greater improvements for hand and foot skin pain NRS, quality of sleep NRS, QoLHEQ score and WPAI overall work impairment and routine activity impairment from baseline to week 16 were seen in the dupilumab group as compared to the placebo group (LS mean change of dupilumab vs placebo: -4.66 vs -1.93 [p<0.0001], 0.88 vs -0.00 [p<0.05], -40.28 vs -16.18 [p<0.0001], -38.57% vs -22.83% [nominal p<0.001] and -36.39% vs -21.26% [nominal p<0.001] respectively).
Clinical efficacy and safety in asthma
The asthma development program included three randomised, double-blind, placebo-controlled, parallel-group, multi-centre studies (DRI12544, QUEST, and VENTURE) of 24 to 52 weeks in treatment duration which enrolled a total of 2,888 patients (12 years of age and older). Patients were enrolled without requiring a minimum baseline blood eosinophil or other type 2 inflammatory biomarkers (e.g. FeNO or IgE) level. Asthma treatment guidelines define type 2 inflammation as eosinophilia ≥150 cells/mcL and/or FeNO ≥20 ppb. In DRI12544 and QUEST, the pre-specified subgroup analyses included blood eosinophils ≥150 and ≥300 cells/mcL, FeNO ≥25 and ≥50 ppb.
DRI12544 was a 24-week dose-ranging study which included 776 patients (18 years of age and older). Dupilumab compared with placebo was evaluated in adult patients with moderate to severe asthma on a medium-to-high dose inhaled corticosteroid and a long acting beta agonist. The primary endpoint was change from baseline to week 12 in FEV1 (L). Annualised rate of severe asthma exacerbation events during the 24-week placebo controlled treatment period was also determined. Results were evaluated in the overall population (unrestricted by minimum baseline eosinophils or other type 2 inflammatory biomarkers) and subgroups based on baseline blood eosinophil count.
QUEST was a 52-week confirmatory study which included 1,902 patients (12 years of age and older). Dupilumab compared with placebo was evaluated in 107 adolescent and 1,795 adult patients with persistent asthma on a medium-to-high dose inhaled corticosteroid (ICS) and a second controller medication. Patients requiring a third controller were allowed to participate in this trial. The primary endpoints were the annualised rate of severe exacerbation events during the 52-week placebo controlled period and change from baseline in pre-bronchodilator FEV1 at week 12 in the overall population (unrestricted by minimum baseline eosinophils or other type 2 inflammatory biomarkers) and subgroups based on baseline blood eosinophil count and FeNO.
VENTURE was a 24-week oral corticosteroid-reduction study in 210 patients with asthma unrestricted by baseline type 2 biomarker levels who required daily oral corticosteroids in addition to regular use of high dose inhaled corticosteroids plus an additional controller. The OCS dose was optimized during the screening period. Patients continued to receive their existing asthma medicine during the study; however their OCS dose was reduced every 4 weeks during the OCS reduction phase (week 4-20), as long as asthma control was maintained. The primary endpoint was the percent reduction in oral corticosteroid dose assessed in the overall population, based on a comparison of the oral corticosteroid dose at weeks 20 to 24 that maintained asthma control with the previously optimized (at baseline) oral corticosteroid dose.
The demographics and baseline characteristics of these 3 studies are provided in Table 15 below.
Table 15. Demographics and baseline characteristics of asthma trials:
| Parameter | DRI12544 (n=776) | QUEST (n=1902) | VENTURE (n=210) |
|---|---|---|---|
| Mean age (years) (SD) | 48.6 (13.0) | 47.9 (15.3) | 51.3 (12.6) |
| % Female | 63.1 | 62.9 | 60.5 |
| % White | 78.2 | 82.9 | 93.8 |
| Duration of Asthma (years), mean ± SD | 22.03 (15.42) | 20.94 (15.36) | 19.95 (13.90) |
| Never smoked, (%) | 77.4 | 80.7 | 80.5 |
| Mean exacerbations in previous year ± SD | 2.17 (2.14) | 2.09 (2.15) | 2.09 (2.16) |
| High dose ICS use (%)a | 49.5 | 51.5 | 88.6 |
| Pre-dose FEV1 (L) at baseline ± SD | 1.84 (0.54) | 1.78 (0.60) | 1.58 (0.57) |
| Mean percent predicted FEV1 at baseline (%)(± SD) | 60.77 (10.72) | 58.43 (13.52) | 52.18 (15.18) |
| % Reversibility (± SD) | 26.85 (15.43) | 26.29 (21.73) | 19.47 (23.25) |
| Mean ACQ-5 score (± SD) | 2.74 (0.81) | 2.76 (0.77) | 2.50 (1.16) |
| Mean AQLQ score (± SD) | 4.02 (1.09) | 4.29 (1.05) | 4.35 (1.17) |
| Atopic Medical History % Overall (AD %, NP %, AR %) | 72.9 (8.0, 10.6, 61.7) | 77.7 (10.3, 12.7, 68.6) | 72.4 (7.6, 21.0, 55.7) |
| Mean FeNO ppb (± SD) | 39.10 (35.09) | 34.97 (32.85) | 37.61 (31.38) |
| % patients with FeNO ppb | |||
| ≥25 | 49.9 | 49.6 | 54.3 |
| ≥50 | 21.6 | 20.5 | 25.2 |
| Mean total IgE IU/mL (± SD) | 435.05 (753.88) | 432.40 (746.66) | 430.58 (775.96) |
| Mean baseline Eosinophil count (± SD) cells/mcL | 350 (430) | 360 (370) | 350 (310) |
| % patients with EOS | |||
| ≥150 cells/mcL | 77.8 | 71.4 | 71.4 |
| ≥300 cells/mcL | 41.9 | 43.7 | 42.4 |
ICS = inhaled corticosteroid; FEV1 = Forced expiratory volume in 1 second; ACQ-5 = Asthma Control Questionnaire-5; AQLQ = Asthma Quality of Life Questionnaire; AD = atopic dermatitis; NP = nasal polyposis; AR = allergic rhinitis; FeNO = fraction of exhaled nitric oxide; EOS = blood eosinophil
a the population in dupilumab asthma trials included patients on medium and high dose ICS. The medium ICS dose was defined as equal to 500 mcg fluticasone or equivalent per day
Exacerbations
In the overall population in DRI12544 and QUEST subjects receiving either dupilumab 200 mg or 300 mg every other week had significant reductions in the rate of severe asthma exacerbations compared to placebo. There were greater reductions in exacerbations in subjects with higher baseline levels of type 2 inflammatory biomarkers such as blood eosinophils or FeNO (Table 16 and Table 17).
Table 16. Rate of severe exacerbations in DRI12544 and QUEST (baseline blood eosinophil levels ≥150 and ≥300 cells/mcL):
| Treatment | Baseline blood EOS | |||||||
|---|---|---|---|---|---|---|---|---|
| ≥150 cells/mcL | ≥300 cells/mcL | |||||||
| Exacerbations per Year | % reduction | Exacerbations per Year | % reduction | |||||
| N | Rate (95% CI) | Rate ratio (95% CI) | N | Rate (95% CI) | Rate ratio (95% CI) | |||
| All Severe Exacerbations | ||||||||
| DRI12544 study | ||||||||
| Dupilumab 200 mg Q2W | 120 | 0.29 (0.16, 0.53) | 0.28a (0.14, 0.55) | 72% | 65 | 0.30 (0.13, 0.68) | 0.29c (0.11, 0.76) | 71% |
| Dupilumab 300 mg Q2W | 129 | 0.28 (0.16, 0.50) | 0.27b (0.14, 0.52) | 73% | 64 | 0.20 (0.08, 0.52) | 0.19d (0.07, 0.56) | 81% |
| Placebo | 127 | 1.05 (0.69, 1.60) | 68 | 1.04 (0.57, 1.90) | ||||
| QUEST study | ||||||||
| Dupilumab 200 mg Q2W | 437 | 0.45 (0.37, 0.54) | 0.44e (0.34.0.58) | 56% | 264 | 0.37 (0.29, 0.48) | 0.34e (0.24.0.48) | 66% |
| Placebo | 232 | 1.01 (0.81, 1.25) | 148 | 1.08 (0.85, 1.38) | ||||
| Dupilumab 300 mg Q2W | 452 | 0.43 (0.36, 0.53) | 0.40e (0.31.0.53) | 60% | 277 | 0.40 (0.32, 0.51) | 0.33e (0.23.0.45) | 67% |
| Placebo | 237 | 1.08 (0.88, 1.33) | 142 | 1.24 (0.97, 1.57) | ||||
ap-value = 0.0003, bp-value = 0.0001, cp-value = 0.0116, dp-value = 0.0024, ep-value < 0.0001 (all statistically significant vs placebo with adjustment for multiplicity); fnominal p-value < 0.0001
Table 17. Rate of severe exacerbations in QUEST defined by baseline FeNO subgroups:
| Treatment | Exacerbations per Year | % reduction | ||
|---|---|---|---|---|
| N | Rate (95% CI) | Rate ratio (95% CI) | ||
| FeNO ≥25 ppb | ||||
| Dupilumab 200 mg Q2W | 299 | 0.35 (0.27, 0.45) | 0.35 (0.25, 0.50)a | 65% |
| Placebo | 162 | 1.00 (0.78, 1.30) | ||
| Dupilumab 300 mg Q2W | 310 | 0.43 (0.35, 0.54) | 0.39 (0.28, 0.54)a | 61% |
| Placebo | 172 | 1.12 (0.88, 1.43) | ||
| FeNO ≥50 ppb | ||||
| Dupilumab 200 mg Q2W | 119 | 0.33 (0.22, 0.48) | 0.31 (0.18, 0.52)a | 69% |
| Placebo | 71 | 1.057 (0.72, 1.55) | ||
| Dupilumab 300 mg Q2W | 124 | 0.39 (0.27, 0.558) | 0.31 (0.19, 0.49)a | 69% |
| Placebo | 75 | 1.27 (0.90, 1.80) | ||
a nominal p-value <0.0001
In the pooled analysis of DRI12544 and QUEST, hospitalisations and/or emergency room visits due to severe exacerbations were reduced by 25.5% and 46.9% with dupilumab 200 mg or 300 mg every other week, respectively.
Lung function
Clinically significant increases in pre-bronchodilator FEV1 were observed at week 12 for DRI12544 and QUEST. There were greater improvements in FEV1 in the subjects with higher baseline levels of type 2 inflammatory biomarkers such as blood eosinophils or FeNO (Table 18 and Table 19).
Significant improvements in FEV1 were observed as early as week 2 following the first dose of dupilumab for both the 200 mg and 300 mg dose strengths and were maintained through week 24 (DRI12544) and week 52 in QUEST (see Figure 3).
Figure 3. Mean change from baseline in pre-bronchodilator FEV1 (L) over time (baseline eosinophils ≥150 and ≥300 cells/mcL and FeNO ≥25 ppb) in QUEST:

Table 18. Mean change from baseline in pre-bronchodilator FEV1 at week 12 in DRI12544 and QUEST (baseline blood eosinophil Levels ≥150 and ≥300 cells/mcL):
| Treatment | Baseline blood EOS | |||||
|---|---|---|---|---|---|---|
| ≥150 cells/mcL | ≥300 cells/mcL | |||||
| N | LS mean Δ from baseline L (%) | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline L (%) | LS mean difference vs. placebo (95% CI) | |
| DRI12544 study | ||||||
| Dupilumab 200 mg Q2W | 120 | 0.32 (18.25) | 0.23a (0.13, 0.33) | 65 | 0.43 (25.9) | 0.26c (0.11, 0.40) |
| Dupilumab 300 mg Q2W | 129 | 0.26 (17.1) | 0.18b (0.08, 0.27) | 64 | 0.39 (25.8) | 0.21d (0.06, 0.36) |
| Placebo | 127 | 0.09 (4.36) | 68 | 0.18 (10.2) | ||
| QUEST study | ||||||
| Dupilumab 200 mg Q2W | 437 | 0.36 (23.6) | 0.17e (0.11, 0.23) | 264 | 0.43 (29.0) | 0.21e (0.13, 0.29) |
| Placebo | 232 | 0.18 (12.4) | 148 | 0.21 (15.6) | ||
| Dupilumab 300 mg Q2W | 452 | 0.37 (25.3) | 0.15e (0.09, 0.21) | 277 | 0.47 (32.5) | 0.24e (0.16, 0.32) |
| Placebo | 237 | 0.22 (14.2) | 142 | 0.22 (14.4) | ||
a p-value < 0.0001, bp-value = 0.0004, cp-value = 0.0008, dp-value = 0.0063, ep-value < 0.0001 (all statistically significant vs placebo with adjustment for multiplicity); fnominal p-value < 0.0001
Table 19. Mean change from baseline in pre-bronchodilator FEV1 at week 12 and week 52 in QUEST by baseline FeNO subgroups:
| Treatment | At week 12 | At week 52 | |||
|---|---|---|---|---|---|
| N | LS mean Δ from baseline L (%) | LS mean difference vs. placebo (95% CI) | LS mean Δ from baseline L (%) | LS mean difference vs. placebo (95% CI) | |
| FeNO ≥25 ppb | |||||
| Dupilumab 200 mg Q2W | 288 | 0.44 (29.0%) | 0.23 (0.15, 0.31)a | 0.49 (31.6%) | 0.30 (0.22, 0.39)a |
| Placebo | 157 | 0.21 (14.1%) | 0.18 (13.2%) | ||
| Dupilumab 300 mg Q2W | 295 | 0.45 (29.8%) | 0.24 (0.16, 0.31)a | 0.45 (30.5%) | 0.23 (0.15, 0.31)a |
| Placebo | 167 | 0.21 (13.7%) | 0.22 (13.6%) | ||
| FeNO ≥50 ppb | |||||
| Dupilumab 200 mg Q2W | 114 | 0.53 (33.5%) | 0.30 (0.17, 0.44)a | 0.59 (36.4%) | 0.38 (0.24, 0.53)a |
| Placebo | 69 | 0.23 (14.9%) | 0.21 (14.6%) | ||
| Dupilumab 300 mg Q2W | 113 | 0.59 (37.6%) | 0.39 (0.26, 0.52)a | 0.55 (35.8%) | 0.30 (0.16, 0.44)a |
| Placebo | 73 | 0.19 (13.0%) | 0.25 (13.6%) | ||
a nominal p-value <0.0001
Quality of life/patient-reported outcomes in asthma
Pre-specified secondary endpoint of ACQ-5 and AQLQ(S) responder rates were analysed at 24 weeks (DRI12544 and VENTURE) and at 52 weeks (QUEST, Table 20). The responder rate was defined as an improvement in score of 0.5 or more (scale range 0-6 for ACQ-5 and 1-7 for AQLQ(S)). Improvements in ACQ-5 and AQLQ(S) were observed as early as week 2 and maintained for 24 weeks in DRI12544 study and 52 weeks in QUEST study. Similar results were observed in VENTURE.
Table 20. ACQ-5 and AQLQ(S) responder rates at week 52 in QUEST:
| PRO | Treatment | EOS ≥150 cells/mcL | EOS ≥300 cells/mcL | FeNO ≥25 ppb | |||
|---|---|---|---|---|---|---|---|
| N | Responder rate % | N | Responder rate (%) | N | Responder rate (%) | ||
| ACQ-5 | Dupilumab 200 mg Q2W | 395 | 72.9 | 239 | 74.5 | 262 | 74.4 |
| Placebo | 201 | 64.2 | 124 | 66.9 | 141 | 65.2 | |
| Dupilumab 300 mg Q2W | 408 | 70.1 | 248 | 71.0 | 277 | 75.8 | |
| Placebo | 217 | 64.5 | 129 | 64.3 | 159 | 64.2 | |
| AQLQ (S) | Dupilumab 200 mg Q2W | 395 | 66.6 | 239 | 71.1 | 262 | 67.6 |
| Placebo | 201 | 53.2 | 124 | 54.8 | 141 | 54.6 | |
| Dupilumab 300 mg Q2W | 408 | 62.0 | 248 | 64.5 | 277 | 65.3 | |
| Placebo | 217 | 53.9 | 129 | 55.0 | 159 | 58.5 | |
Oral corticosteroid reduction study (VENTURE)
VENTURE evaluated the effect of dupilumab on reducing the use of maintenance oral corticosteroids. Baseline characteristics are presented in Table 15. All patients were on oral corticosteroids for at least 6 months prior to the study initiation. The baseline mean oral corticosteroid use was 11.75 mg in the placebo group and 10.75 mg in the group receiving dupilumab.
In this 24-week trial, asthma exacerbations (defined as a temporary increase in oral corticosteroid dose for at least 3 days) were reduced by 59% in subjects receiving dupilumab compared with those receiving placebo (annualised rate 0.65 and 1.60 for the dupilumab and placebo group, respectively; rate ratio 0.41 [95% CI 0.26, 0.63]) and improvement in pre-bronchodilator FEV1 from baseline to week 24 was greater in subjects receiving dupilumab compared with those receiving placebo (LS mean difference for dupilumab versus placebo of 0.22 L [95% CI: 0.09 to 0.34 L]). Effects on lung function, on oral steroid and exacerbation reduction were similar irrespective of baseline levels of type 2 inflammatory biomarkers (e.g. blood eosinophils, FeNO). The ACQ-5 and AQLQ(S) were also assessed in VENTURE and showed improvements similar to those in QUEST.
The results for VENTURE by baseline biomarkers are presented in the Table 21.
Table 21. Effect of dupilumab on OCS dose reduction, VENTURE (baseline blood eosinophil levels ≥150 and ≥300 cells/mcL and FeNO ≥25 ppb):
| Baseline blood EOS ≥150 cells/mcL | Baseline blood EOS ≥300 cells/mcL | FeNO ≥25 ppb | ||||
|---|---|---|---|---|---|---|
| Dupilumab 300 mg Q2W N=81 | Placebo N=69 | Dupilumab 300 mg Q2W N=48 | Placebo N=41 | Dupilumab 300 mg Q2W N=57 | Placebo N=57 | |
| Primary endpoint (week 24) | ||||||
| Percent reduction in OCS from baseline | ||||||
| Mean overall percent reduction from baseline (%) Difference (% [95% CI]) (Dupilumab vs. placebo) | 75.91 29.39b (15.67, 43.12) | 46.51 | 79.54 36.83b (18.94, 54.71) | 42.71 | 77.46 34.53b (19.08, 49.97) | 42.93 |
| Median % reduction in daily OCS dose from baseline | 100 | 50 | 100 | 50 | 100 | 50 |
| Percent reduction from baseline 100% ≥90% ≥75% ≥50% >0% No reduction or any increase in OCS dose, or dropped out of study | 54.3 58.0 72.8 82.7 87.7 12.3 | 33.3 34.8 44.9 55.1 66.7 33.3 | 60.4 66.7 77.1 85.4 85.4 14.6 | 31.7 34.1 41.5 53.7 63.4 36.6 | 52.6 54.4 73.7 86.0 89.5 10.5 | 28.1 29.8 36.8 50.9 66.7 33.3 |
| Secondary endpoint (week 24)a | ||||||
| Proportion of patients achieving a reduction of OCS dose to <5 mg/day | 77 | 44 | 84 | 40 | 79 | 34 |
| Odds ratio (95% CI) | 4.29c (2.04, 9.04) | 8.04d (2.71, 23.82) | 7.21b (2.69, 19.28) | |||
a model estimates by logistic regression, bnominal p-value < 0.0001, cnominal p-value = 0.0001, dnominal p-value = 0.0002
Long-term extension study (TRAVERSE)
The long-term safety of dupilumab in 2,193 adults and 89 adolescents with moderate-to-severe asthma, including 185 adults with oral corticosteroid-dependent asthma, who had participated in previous clinical trials of dupilumab (DRI12544, QUEST, and VENTURE), was assessed in the open-label extension study (TRAVERSE) (see section 4.8). Efficacy was measured as a secondary endpoint, was similar to results observed in the pivotal studies and was sustained up to 96 weeks. In the adults with oral-corticosteroid-dependent asthma, there was sustained reduction in exacerbations and improvement in lung function up to 96 weeks, despite decrease or discontinuation of oral corticosteroid dose.
Paediatric study (6 to 11 years of age; VOYAGE)
The efficacy and safety of dupilumab in paediatric patients was evaluated in a 52-week multicentre, randomised, double-blind, placebo-controlled study (VOYAGE) in 408 patients 6 to 11 years of age, with moderate-to-severe asthma on a medium- or high-dose ICS and one controller medication or high dose ICS alone. Patients were randomised to dupilumab (N=273) or matching placebo (N=135) every other week based on body weight ≤30 kg or >30 kg, respectively. The efficacy was evaluated in populations with type 2 inflammation defined as blood eosinophil levels of ≥150 cells/mcL or FeNO ≥20 ppb.
The primary endpoint was the annualised rate of severe exacerbation events during the 52-week placebo-controlled period and the key secondary endpoint was the change from baseline in pre-bronchodilator FEV1 percent predicted at week 12. Additional secondary endpoints included mean change from baseline and responder rates in the ACQ-7-IA and PAQLQ(S)-IA scores.
The demographics and baseline characteristics for VOYAGE are provided in Table 22 below.
Table 22. Demographics and baseline characteristics for VOYAGE:
| Parameter | EOS ≥150 cells/mcL or FeNO ≥20 ppb (N=350) | EOS ≥300 cells/mcL (N=259) |
|---|---|---|
| Mean age (years) (SD) | 8.9 (1.6) | 9.0 (1.6) |
| % Female | 34.3 | 32.8 |
| % White | 88.6 | 87.3 |
| Mean body weight (kg) | 36.09 | 35.94 |
| Mean exacerbations in previous year (± SD) | 2.47 (2.30) | 2.64 (2.58) |
| ICS dose (%) Medium High | 55.7 43.4 | 54.4 44.4 |
| Pre-dose FEV1 (L) at baseline (± SD) | 1.49 (0.41) | 1.47 (0.42) |
| Mean percent predicted FEV1 (%) (±SD) | 77.89 (14.40) | 76.85 (14.78) |
| Mean % Reversibility (± SD) | 27.79 (19.34) | 22.59 (20.78) |
| Mean ACQ-7-IA score (± SD) | 2.14 (0.72) | 2.16 (0.75) |
| Mean PAQLQ(S)-IA score (± SD) | 4.94 (1.10) | 4.93 (1.12) |
| Atopic Medical History % Overall (AD %, AR %) | 94 (38.9, 82.6) | 96.5 (44.4, 85.7) |
| Median total IgE IU/mL (± SD) | 905.52 (1140.41) | 1077.00 (1230.83) |
| Mean FeNO ppb (± SD) | 30.71 (24.42) | 33.50 (25.11) |
| % patients with FeNO ≥20 ppb | 58 | 64.1 |
| Mean baseline Eosinophil count (± SD) cells/mcL | 570 (380) | 710 (360) |
| % patients with EOS ≥150 cells/mcL ≥300 cells/mcL | 94.6 74 | 0 100 |
ICS = inhaled corticosteroid; FEV1 = Forced expiratory volume in 1 second; ACQ-7-IA = Asthma Control Questionnaire-7 Interviewer Administered; PAQLQ(S)-IA = Paediatric Asthma Quality of Life Questionnaire with Standardised Activities–Interviewer Administered; AD = atopic dermatitis; AR = allergic rhinitis; EOS = blood eosinophil; FeNO = fraction of exhaled nitric oxide
Dupilumab significantly reduced the annualised rate of severe asthma exacerbation events during the 52-week treatment period compared to placebo in the population with the type 2 inflammation and in population defined by baseline blood eosinophils ≥300 cells/mcL or by baseline FeNO ≥20 ppb. Clinically significant improvements in percent predicted pre-bronchodilator FEV1 were observed at week 12. Improvements were also observed for ACQ-7-IA and PAQLQ(S)-IA at week 24 and were sustained at week 52. Greater responder rates were observed for ACQ-7-IA and PAQLQ(S)-IA compared to placebo at week 24. The efficacy results for VOYAGE are presented in Table 21.
In the population with the type 2 inflammation, the LS mean change from baseline in prebronchodilator FEV1 at week 12 was 0.22 L in the dupilumab group and 0.12 L in the placebo group, with an LS mean difference versus placebo of 0.10 L (95% CI: 0.04, 0.16). The treatment effect was sustained over the 52-week treatment period, with an LS mean difference versus placebo at week 52 of 0.17 L (95% CI: 0.09, 0.24).
In the population defined by baseline blood eosinophils ≥300 cells/mcL, the LS mean change from baseline in pre-bronchodilator FEV1 at week 12 was 0.22 L in the dupilumab group and 0.12 L in the placebo group, with an LS mean difference versus placebo of 0.10 L (95% CI: 0.03, 0.17). The treatment effect was sustained over the 52-week treatment period, with an LS mean difference versus placebo at week 52 of 0.17 L (95% CI: 0.09, 0.26).
In both primary efficacy populations, there was a rapid improvement in FEF25-75% and FEV1/FVC (onset of a difference was observed as early as week 2) and sustained over the 52-week treatment period, see Table 23.
Table 23. Rate of severe exacerbations, mean change from baseline in FEV1. ACQ-7-IA and PAQLQ(S)-IA responder rates in VOYAGE:
| Treatment | EOS ≥150 cells/mcL or FeNO ≥20 ppb | EOS ≥300 cells/mcL | FeNO ≥20 ppb | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Annualised severe exacerbations rate over 52 weeks | |||||||||
| N | Rate (95% CI) | Rate ratio (95% CI) | N | Rate (95% CI) | Rate ratio (95% CI) | N | Rate (95% CI) | Rate ratio (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 236 | 0.305 (0.223, 0.416) | 0.407b (0.274, 0.605) | 175 | 0.235 (0.160, 0.345) | 0.353b (0.222, 0.562) | 141 | 0.271 (0.170, 0.432) | 0.384c (0.227, 0.649) |
| Placebo | 114 | 0.748 (0.542, 1.034) | 84 | 0.665 (0.467, 0.949) | 62 | 0.705 (0.421, 1.180) | |||
| Mean change from baseline in percent predicted FEV1 at week 12 | |||||||||
| N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 229 | 10.53 | 5.21c (2.14, 8.27) | 168 | 10.15 | 5.32d (1.76, 8.88) | 141 | 11.36 | 6.74d (2.54, 10.93) |
| Placebo | 110 | 5.32 | 80 | 4.83 | 62 | 4.62 | |||
| Mean change from baseline in percent predicted FEF 25-75% at week 12 | |||||||||
| N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 229 | 16.70 | 11.93e (7.44, 16.43) | 168 | 16.91 | 13.92e (8.89, 18.95) | 141 | 17.96 | 13.97e (8.30, 19.65) |
| Placebo | 110 | 4.76 | 80 | 2.99 | 62 | 3.98 | |||
| Mean change from baseline in FEV1/FVC % at week 12 | |||||||||
| N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | N | LS mean Δ from baseline | LS mean difference vs. placebo (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 229 | 5.67 | 3.73e (2.25, 5.21) | 168 | 6.10 | 4.63e (2.97, 6.29) | 141 | 6.84 | 4.95e (3.08, 6.81) |
| Placebo | 110 | 1.94 | 80 | 1.47 | 62 | 1.89 | |||
| ACQ-7-IA at week 24a | |||||||||
| N | Responder rate % | OR vs. placebo (95% CI) | N | Responder rate % | OR vs. placebo (95% CI) | N | Responder rate % | OR vs. placebo (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 236 | 79.2 | 1.82g (1.02, 3.24) | 175 | 80.6 | 2.79f (1.43, 5.44) | 141 | 80.9 | 2.60g (1.21, 5.59) |
| Placebo | 114 | 69.3 | 84 | 64.3 | 62 | 66.1 | |||
| PAQLQ(S)-IA at week 24a | |||||||||
| N | Responder rate % | OR vs. placebo (95% CI) | N | Responder rate % | OR vs. placebo (95% CI) | N | Responder rate % | OR vs. placebo (95% CI) | |
| Dupilumab 100 mg Q2W (<30 kg)/ 200 mg Q2W (≥30 kg) | 211 | 73.0 | 1.57 (0.87, 2.84) | 158 | 72.8 | 1.84 (0.92, 3.65) | 131 | 75.6 | 2.09 (0.95, 4.61) |
| Placebo | 107 | 65.4 | 81 | 63.0 | 61 | 67.2 | |||
a the responder rate was defined as an improvement in score of 0.5 or more (scale range 0-6 for ACQ-7-IA and 1-7 for PAQLQ(S))
b p-value < 0.0001; cp-value < 0.001, dp-value < 0.01 (all statistically significant vs placebo with adjustment for multiplicity); enominal p-value < 0.0001, fnominal p-value < 0.01, gnominal p-value < 0.05
Significant improvements in percent predicted FEV1 were observed as early as week 2 and were maintained through week 52 in VOYAGE study.
Improvements in percent predicted FEV1 over time in VOYAGE are shown in Figure 4.
Figure 4. Mean change from baseline in percent predicted pre-bronchodilator FEV1 (L) over time in VOYAGE (baseline blood eosinophils ≥150 cells/mcL or FeNO ≥20 ppb, baseline eosinophils ≥300 cells/mcL, and baseline FeNO ≥20 ppb):

In VOYAGE, in the population with the type 2 inflammation, the mean annualised total number of systemic corticosteroid courses due to asthma was reduced by 59.3% versus placebo (0.350 [95% CI: 0.256, 0.477] versus 0.860 [95% CI: 0.616, 1.200]). In the population defined by baseline blood eosinophils ≥300 cells/mcL, the mean annualised total number of systemic corticosteroid courses due to asthma was reduced by 66.0% versus placebo (0.274 [95% CI: 0.188, 0.399] versus 0.806 [95% CI: 0.563, 1.154]).
Dupilumab improved the overall health status as measured by the European Quality of Life 5-Dimension Youth Visual Analog Scale (EQ-VAS) in both the type 2 inflammation and the baseline blood eosinophil count of ≥300 cells/mcL populations at week 52; the LS mean difference versus placebo was 4.73 (95% CI: 1.18, 8.28), and 3.38 (95% CI: -0.66, 7.43), respectively.
Dupilumab reduced the impact of paediatric patient's asthma on the caregiver quality of life as measured by the Paediatric Asthma Quality of Life Questionnaire (PACQLQ) in both the type 2 inflammation and the baseline blood eosinophil count of ≥300 cells/mcL population at week 52; the LS mean difference versus placebo was 0.47 (95% CI: 0.22, 0.72), and 0.50 (95% CI: 0.21, 0.79), respectively.
Long-term extension study (EXCURSION)
The efficacy of dupilumab, measured as a secondary endpoint, was assessed in 365 paediatric asthma patients (6 to 11 years of age) in the long-term extension study (EXCURSION). There were sustained reductions in exacerbations requiring hospitalization and/or emergency room visits and a reduction in exposure to systemic oral corticosteroids. Sustained improvements in lung function were observed across multiple parameters including percent predicted FEV1. percent predicted FVC, FEV1/FVC ratio and percent predicted FEF 25-75%. Furthermore, 75% of patients achieved and/or maintained normal lung function with pre-bronchodilator percent predicted FEV1 > 80% by the end of EXCURSION. Efficacy was sustained for a cumulative treatment duration of up to 104 weeks (VOYAGE and EXCURSION).
Clinical efficacy in chronic rhinosinusitis with nasal polyposis (CRSwNP)
The chronic rhinosinusitis with nasal polyposis (CRSwNP) development program included two randomised, double-blind, parallel-group, multicentre, placebo-controlled studies (SINUS-24 and SINUS-52) in 724 patients aged 18 years and older on background intranasal corticosteroids (INCS). These studies included patients with severe CRSwNP despite prior sino-nasal surgery or treatment with, or who were ineligible to receive, systemic corticosteroids in the past 2 years. Rescue with systemic corticosteroids or surgery was allowed during the studies at the investigator's discretion All patients had evidence of sinus opacification on the Lund MacKay (LMK) sinus CT scan and 73% to 90% of patients had opacification of all sinuses. Patients were stratified based on their histories of prior surgery and co-morbid asthma/nonsteroidal anti-inflammatory drug exacerbated respiratory disease (NSAID-ERD).
The co-primary efficacy endpoints were change from baseline to week 24 in bilateral endoscopic nasal polyps score (NPS) as graded by central blinded readers, and change from baseline to week 24 in nasal congestion/obstruction score averaged over 28 days (NC), as determined by patients using a daily diary. For NPS, polyps on each side of the nose were graded on a categorical scale (0=no polyps; 1=small polyps in the middle meatus not reaching below the inferior border of the middle turbinate; 2=polyps reaching below the lower border of the middle turbinate; 3=large polyps reaching the lower border of the inferior turbinate or polyps medial to the middle turbinate; 4=large polyps causing complete obstruction of the inferior nasal cavity). The total score was the sum of the right and left scores. Nasal congestion was rated daily by the subjects on a 0 to 3 categorical severity scale (0=no symptoms; 1=mild symptoms; 2=moderate symptoms; 3=severe symptoms).
The demographics and baseline characteristics of these 2 studies are provided in Table 24 below.
Table 24. Demographics and baseline characteristics of CRSwNP studies:
| Parameter | SINUS-24 (N=276) | SINUS-52 (N=448) |
|---|---|---|
| Mean age (years) (SD) | 50.49 (13.39) | 51.95 (12.45) |
| % Male | 57.2 | 62.3 |
| Mean CRSwNP duration (years)(SD) | 11.11 (9.16) | 10.94 (9.63) |
| Patients with ≥1 prior surgery (%) | 71.7 | 58.3 |
| Patients with systemic corticosteroid use in the previous 2 years (%) | 64.9 | 80.1 |
| Mean Bilateral endoscopic NPSa (SD), range 0–8 | 5.75 (1.28) | 6.10 (1.21) |
| Mean Nasal congestion (NC) scorea (SD) range 0–3 | 2.35 (0.57) | 2.43 (0.59) |
| Mean LMK sinus CT total scorea (SD), range 0–24 | 19.03 (4.44) | 17.96 (3.76) |
| Mean Smell test (UPSIT) scorea (SD), range 0–40 | 14.56 (8.48) | 13.61 (8.02) |
| Mean loss of smell scorea (AM), (SD) range 0–3 | 2.71 (0.54) | 2.75 (0.52) |
| Mean SNOT-22 total scorea (SD), range 0–110 | 49.40 (20.20) | 51.86 (20.90) |
| Mean Rhinosinusitis severity scalea (VAS), (SD) 0–10 cm | 7.68 (2.05) | 8.00 (2.08) |
| Mean blood eosinophils (cells/mcL)(SD) | 437 (333) | 431 (353) |
| Mean total IgE IU/mL (SD) | 211.97 (275.73) | 239.84 (341.53) |
| Atopic (type 2 inflammatory disease) Medical History % Overall | 75.4% | 82.4% |
| Asthma (%) | 58.3 | 59.6 |
| Mean FEV1 (L)(SD) | 2.69 (0.96) | 2.57 (0.83) |
| Mean FEV1 percent predicted (%)(SD) | 85.30 (20.23) | 83.39 (17.72) |
| Mean ACQ-6 scorea (SD) | 1.62 (1.14) | 1.58 (1.09) |
| NSAID-ERD (%) | 30.4 | 26.8 |
a higher scores indicate greater disease severity except UPSIT where higher scores indicate lower disease severity; SD=standard deviation; AM = morning; NPS = nasal polyps score; UPSIT = University of Pennsylvania smell identification test; SNOT-22 = 22-item Sino-Nasal Outcome Test; VAS = visual analogue scale; FEV1 = Forced expiratory volume in 1 second; ACQ-6 = Asthma Control Questionnaire-6; NSAID-ERD = aspirin/nonsteroidal anti-inflammatory drug exacerbated respiratory disease
Clinical Response (SINUS-24 and SINUS-52)
The results for primary and secondary endpoints in CRSwNP studies are presented in the Table 25.
Table 25. Results of the primary and secondary endpoints in CRSwNP trials:
| SINUS-24 | SINUS-52 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n=133) | Dupilumab 300 mg Q2W (n=143) | LS mean difference vs. placebo (95%CI) | Placebo (n=153) | Dupilumab 300 mg Q2W (n=295) | LS mean difference vs. placebo (95%CI) | |||||
| Primary endpoints at week 24 | ||||||||||
| Scores | Baseline mean | LS mean change | Baseline mean | LS mean change | Baseline mean | LS mean change | Baseline mean | LS mean change | ||
| NPS | 5.86 | 0.17 | 5.64 | -1.89 | -2.06 (-2.43. -1.69) | 5.96 | 0.10 | 6.18 | -1.71 | -1.80 (-2.10. -1.51) |
| NC | 2.45 | -0.45 | 2.26 | -1.34 | -0.89 (-1.07. -0.71) | 2.38 | -0.38 | 2.46 | -1.25 | -0.87 (-1.03. -0.71) |
| Key secondary endpoints at week 24 | ||||||||||
| Scores | Baseline mean | LS mean change | Baseline mean | LS mean change | Baseline mean | LS mean change | Baseline mean | LS mean change | ||
| LMK sinus CT scan score | 19.55 | -0.74 | 18.55 | -8.18 | -7.44 (-8.35. -6.53) | 17.65 | -0.09 | 18.12 | -5.21 | -5.13 (-5.80. -4.46) |
| Total symptom score | 7.28 | -1.17 | 6.82 | -3.77 | -2.61 (-3.04. -2.17) | 7.08 | -1.00 | 7.30 | -3.45 | -2.44 (-2.87. -2.02) |
| UPSIT | 14.44 | 0.70 | 14.68 | 11.26 | 10.56 (8.79, 12.34) | 13.78 | -0.81 | 13.53 | 9.71 | 10.52 (8.98, 12.07) |
| Loss of smell | 2.73 | -0.29 | 2.70 | -1.41 | -1.12 (-1.31. -0.93) | 2.72 | -0.23 | 2.77 | -1.21 | -0.98 (-1.15. -0.81) |
| SNOT-22 | 50.87 | -9.31 | 48.0 | -30.43 | -21.12 (-25.17. -17.06) | 53.48 | -10.40 | 51.02 | -27.77 | -17.36 (-20.87. -13.85) |
| VAS | 7.96 | -1.34 | 7.42 | -4.54 | -3.20 (-3.79. -2.60) | 7.98 | -1.39 | 8.01 | -4.32 | -2.93 (-3.45. -2.40) |
A reduction in score indicates improvement, except UPSIT where an increase indicates improvement. Total symptom score is a composite severity score consisting of the sum of daily symptoms of NC, loss of smell, and anterior/posterior rhinorrhoea. NC = nasal congestion, NPS = nasal polyposis score; LMK = Lund-MacKay total CT score; UPSIT = University of Pennsylvania smell identification test; SNOT-22 = 22-item Sino-Nasal Outcome Test; TSS = total symptom score; VAS = visual analogue scale for rhinosinusitis (all p-values < 0.0001 (all statistically significant vs placebo with adjustment for multiplicity); nominal for VAS)
The results of SINUS-52 study at week 52 are presented in Table 26.
Table 26. Results of the efficacy at week 52 in SINUS-52 study:
| Placebo (n=153) | Dupilumab 300 mg Q2W (n=150) | LS mean difference vs. placebo (95% CI) | Dupilumab 300 mg Q2W-Q4W (n=145) | LS mean difference vs. placebo (95%CI) | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline mean | LS mean change | Baseline mean | LS mean change | Baseline mean | LS mean change | |||
| NPS | 5.96 | 0.15 | 6.07 | -2.24 | -2.40a (-2.77. -2.02) | 6.29 | -2.06 | -2.21b (-2.59. -1.83) |
| NC | 2.38 | -0.37 | 2.48 | -1.35 | -0.98a (-1.17. -0.79) | 2.44 | -1.48 | -1.10b (-1.29. -0.91) |
| LMK sinus CT scan score | 17.65 | 0.11 | 18.42 | -6.83 | -6.94b (-7.87. -6.01) | 17.81 | -5.60 | -5.71b (-6.64. -4.77) |
| Total symptom score | 7.08 | -0.94 | 7.31 | -3.79 | -2.85b (-3.35. -2.35) | 7.28 | -4.16 | -3.22b (-3.73. -2.72) |
| UPSIT | 13.78 | -0.77 | 13.46 | 9.53 | 10.30b (8.50, 12.10) | 13.60 | 9.99 | 10.76b (8.95, 12.57) |
| Loss of Smell | 2.72 | -0.19 | 2.81 | -1.29 | -1.10b (-1.31. -0.89) | 2.73 | -1.49 | -1.30b (-1.51. -1.09) |
| SNOT-22 | 53.48 | -8.88 | 50.16 | -29.84 | -20.96a (-25.03. -16.89) | 51.89 | -30.52 | -21.65b (-25.71. -17.58) |
| VAS | 7.98 | -0.93 | 8.24 | -4.74 | -3.81b (-4.46. -3.17) | 7.78 | -4.39 | -3.46b (-4.10. -2.81) |
A reduction in score indicates improvement, except UPSIT where an increase indicates improvement. Total symptom score is a composite severity score consisting of the sum of daily symptoms of NC, loss of smell, and anterior/posterior rhinorrhoea. NC = nasal congestion, NPS = nasal polyposis score; LMK = Lund-MacKay total CT score; UPSIT = University of Pennsylvania smell identification test; SNOT-22 = 22-item Sino-Nasal Outcome Test; TSS = total symptom score; VAS = visual analogue scale for rhinosinusitis ( a p-value < 0.0001 (all statistically significant vs placebo with adjustment for multiplicity); b nominal pvalue <0.0001
Statistically significant and clinically meaningful efficacy was observed in SINUS-24 with regard to improvement in bilateral endoscopic NPS score at week 24. In the post-treatment period when patients were off dupilumab, the treatment effect diminished over time (see Figure 5a). Similar results were also seen in SINUS-52 at both week 24 and week 52 with a progressive improvement over time (see Figure 5b).
Figure 5. LS mean change from baseline in bilateral nasal polyps score (NPS) in SINUS-24 and SINUS-52 - ITT population:

In both studies, significant improvements in NC and daily loss of smell severity were observed as early as the first assessment at week 4. The LS mean difference for NC at week 4 in the dupilumab group versus placebo was -0.41 (95% CI: -0.52. -0.30) in SINUS-24 and -0.37 (95% CI: -0.46. -0.27) in SINUS-52. The LS mean difference for loss of smell at week 4 in the dupilumab group versus placebo was -0.34 (95% CI: -0.44. -0.25) in SINUS-24 and -0.31 (95% CI: -0.41. -0.22) in SINUS-52. A reduction in the proportion of patients with anosmia was observed in SINUS-24 and SINUS-52. At baseline, 74% to 79% of patients had anosmia, which was reduced to 24% in SINUS-24 and 30% in SINUS-52 at week 24. compared to no change in placebo. Improvement in nasal peak inspiratory flow (NPIF) was observed in SINUS-24 and SINUS-52 at week 24. The LS mean difference in the dupilumab group versus placebo was 40.4 L/min (95% CI: 30.4, 50.4) and 36.6 L/min (95% CI: 28.0, 45.3), respectively.
Among the patients with rhinosinusitis VAS score >7 at baseline, a higher percentage of patients achieved VAS ≤7 in the dupilumab group compared with the placebo group (83.3% versus 39.4% in SINUS-24 and 75.0% versus 39.3% in SINUS-52) at week 24.
In the pre-specified multiplicity-adjusted pooled analysis of two studies, treatment with dupilumab resulted in significant reduction of systemic corticosteroid use and need for sino-nasal surgery versus placebo (HR of 0.24; 95% CI: 0.17, 0.35) (see Figure 6). The proportion of patients who required systemic corticosteroids was reduced by 74% (HR of 0.26; 95% CI: 0.18, 0.38). The total number of systemic corticosteroid courses per year was reduced by 75% (RR of 0.25; 95% CI: 0.17, 0.37). The mean individual annualised prescribed total dose of systemic corticosteroids (in mg) during the treatment period was 71% lower in the pooled dupilumab group compared with the pooled placebo group (60.5 [531.3] mg versus 209.5 [497.2] mg, respectively). The proportion of patients who required surgery was reduced by 83% (HR of 0.17; 95% CI: 0.07, 0.46).
Figure 6. Kaplan Meier Curve for time to first systemic corticosteroid use and/or sino-nasal surgery during treatment period - ITT population [SINUS-24 and SINUS-52 pooled]:

The effects of dupilumab on the primary endpoints of NPS and nasal congestion and the key secondary endpoint of LMK sinus CT scan score were consistent in patients with prior surgery and without prior surgery.
In patients with co-morbid asthma, significant improvements in FEV1 and ACQ-6 were observed at week 24 irrespective of baseline blood eosinophil levels. The pooled LS Mean change from baseline in FEV1 at week 24 for dupilumab 300 mg Q2W was 0.14 vs -0.07 L for placebo, for a difference of 0.21 L (95% CI: 0.13, 0.29). In addition, improvements in FEV1 were noted from the first post-baseline assessment, at week 8 in SINUS-24 and week 4 in SINUS-52. Improvements in ACQ-6 in patients with co-morbid asthma were observed in both studies. A response was defined as an improvement in score of 0.5 or more. The LS mean difference in the dupilumab group versus placebo at week 24 was -0.76 (95% CI: -1.00 to -0.51) in SINUS-24 and -0.94 (95% CI: -1.19, -0.69) in SINUS-52.
The ACQ-6 responder rate for dupilumab 300 mg Q2W for SINUS-24 at week 24 was 56% versus 28% in placebo (odds ratio 3.17; 95% CI: 1.65, 6.09). The ACQ-6 responder rate for dupilumab 300 mg Q2W for SINUS-52 was 46% versus 14% placebo at week 52 (odds ratio 7.02; 95% CI: 3.10, 15.90).
In patients with NSAID-ERD, the effects of dupilumab on the primary endpoints of NPS and NC and the key secondary endpoint of LMK sinus CT scan score were consistent with that observed in the overall CRSwNP population.
Clinical efficacy in prurigo nodularis (PN)
The prurigo nodularis (PN) development program included two 24-week randomised, double-blind, placebo-controlled, multicenter, parallel-group studies (PRIME and PRIME2) in 311 patients 18 years of age and older with moderate to severe PN, defined as severe pruritus (WI-NRS ≥7 on a scale of 0 to 10) and greater than or equal to 20 nodular lesions, whose disease was not adequately controlled with topical prescription therapies or when those therapies were not advisable. PRIME and PRIME2 assessed the effect of dupilumab on itch improvement as well as its effect on PN lesions, Dermatology Life Quality Index (DLQI), Hospital Anxiety and Depression Scale (HADS) and skin pain.
In these two studies, patients received either subcutaneous dupilumab 600 mg (two 300 mg injections) on day 1, followed by 300 mg once every other week (Q2W) for 24 weeks, or matching placebo.
In these studies, the mean age was 49.5 years, the median weight was 71.3 kg, 65.3% of patients were female, 56.6% were White, 6.1% were Black, and 34.1% were Asian. At baseline, the mean WI-NRS was 8.5, 66.3% had 20 to 100 nodules (moderate), 33.7% had greater than 100 nodules (severe), 99.7% received prior topical therapies, 12.5% received prior systemic corticosteroids, 20.6% received prior systemic non-steroidal immunosuppressants, and 4.5% prior gabapentinoids. Eleven percent of patients were taking stable doses of antidepressants at baseline and were instructed to continue taking these medications during the study. 43.4% had history of atopy (defined as having a medical history of AD, allergic rhinitis/rhinoconjunctivitis, asthma, or food allergy).
The WI-NRS is comprised of a single item, rated on a scale from 0 ("no itch") to 10 ("worst imaginable itch"). Participants were asked to rate the intensity of their worst pruritus (itch) over the past 24 hours using this scale. The IGA PN-S is a scale that measures the approximate number of nodules using a 5-point scale from 0 (clear) to 4 (severe).
The primary efficacy endpoint was the proportion of patients with improvement (reduction) in WI-NRS by ≥4. Key secondary endpoints included the proportion of participants with IGA PN-S 0 or 1 (the equivalent of 0-5 nodules).
The efficacy results for PRIME and PRIME2 are presented in Table 27 and Figures 7 and 8.
Table 27. Results of the Primary and Secondary Endpoints in PRIME and PRIME2:
| PRIME | PRIME2 | |||||
|---|---|---|---|---|---|---|
| Placebo (N=76) | Dupilumab 300 mg Q2W (N=75) | Difference (95% CI) for Dupilumab vs. Placebo | Placebo (N=82) | Dupilumab 300 mg Q2W (N=78) | Difference (95% CI) for Dupilumab vs. Placebo | |
| Proportion of patients with improvement (reduction) in WI- NRS by ≥4 points from baseline at week 24 (Primary endpoint in PRIME)b | 18.4% | 60.0% | 42.7% (27.76, 57.72) | 19.5% | 57.7% | 42.6% (29.06, 56.08) |
| Proportion of patients with improvement (reduction) in WI- NRS by ≥4 points from baseline at week 12. (Primary endpoint in PRIME2)b | 15.8%a | 44.0%a | 29.2% (14.49, 43.81)a | 22.0% | 37.2% | 16.8% (2.34, 31.16) |
| Proportion of patients with IGA PN-S 0 or 1 at week 24.b | 18.4% | 48.0% | 28.3% (13.41, 43.16) | 15.9% | 44.9% | 30.8% (16.37, 45.22) |
| Proportion of patients with both an improvement (reduction) in WI- NRS by ≥4 points from baseline to Week 24 and an IGA PN-S 0 or 1 at Week 24b | 9.2% | 38.7% | 29.6% (16.42, 42.81) | 8.5% | 32.1% | 25.5% (13.09, 37.86) |
| % change from baseline in WI- NRS at week 24 (SE) | -22.22 (5.74) | -48.89 (5.61) | -26.67 (-38.44, -14.90) | -36.18 (6.21) | -59.34 (6.39) | -23.16 (-33.81, -12.51) |
| Change from baseline in DLQI at week 24 (SE) | -5.77 (1.05) | -11.97 (1.02) | -6.19 (-8.34, -4.05) | -6.77 (1.18) | -13.16 (1.21) | -6.39 (-8.42, -4.36) |
| Change from baseline in skin pain- NRS at week 24 (SE)c | -2.16 (0.44) | -4.33 (0.43) | -2.17 (-3.07, -1.28) | -2.74 (0.51) | -4.35 (0.53) | -1.61 (-2.49, -0.73) |
| Change from baseline in HADS at week 24 (SE)c | -2.02 (0.94) | -4.62 (0.93) | -2.60 (-4.52, -0.67) | -2.59 (1.03) | -5.55 (1.06) | -2.96 (-4.73, -1.19) |
a Not adjusted for multiplicity in PRIME.
b Subjects who received rescue treatment earlier or had missing data were considered as non-responders.
c Subjects who received rescue treatment earlier or discontinued due to lack of efficacy were imputed using worst observation carried forward; other missing data were imputed using multiple imputation.
SE = secondary endpoint
The onset of action in change from baseline in WI-NRS, defined as the first timepoint at which difference from placebo was and remained significant (nominal p<0.05) in the weekly average of daily WI-NRS, was observed as early as Week 3 in PRIME (Figure 7a) and Week 4 in PRIME2 (Figure7b).
Figure 7. LS mean percent change from baseline in WI-NRS in PRIME and PRIME2 up to Week 24:

A greater proportion of patients experienced WI-NRS improvements of ≥4 points from baseline by Weeks 4 and 11 in the dupilumab group as compared to the placebo group in PRIME (Figure 8a nominal p<0.007) and PRIME2 (Figure 8b nominal p<0.013), respectively, and this difference remained significant throughout the treatment period.
Figure 8. Proportion of patients with WI-NRS ≥4 improvement over time in PRIME and PRIME2:

Treatment effects in subgroups (age, gender, with or without medical history of atopy, and background treatment, including immunosuppressants) in PRIME and PRIME2 were consistent with the results in the overall study population.
Once treatment was discontinued after 24 weeks, there was an indication towards recurrence of signs and symptoms within the 12-week follow-up period.
Clinical efficacy in eosinophilic esophagitis (EoE)
Adult and Paediatric Patients 12 to 17 Years of Age with EoE
The eosinophilic esophagitis (EoE) development program included a three-part protocol up to 52-weeks (TREET) consisting of two separately randomised, double-blind, parallel-group, multicentre, placebo-controlled, 24-week treatment studies (TREET Part A and TREET Part B) followed by a 28 week active treatment extension study (TREET C) in adult and paediatric patients 12 to 17 years of age, excluding patients <40 kg. In TREET Parts A and B, all enrolled patients had to have failed conventional medicinal therapy (proton pump inhibitors), 74% were treated with another conventional medicinal therapy (swallowed topical corticosteroids) prior to inclusion. In TREET Part B, 49% of patients were inadequately controlled, intolerant or contraindicated to swallowed topical corticosteroid treatment. In both parts, patients were required to have ≥15 intraepithelial eosinophils per high-power field (eos/hpf) following an at least 8-week course of a high-dose proton pump inhibitor (PPI) either prior to or during the screening period and a Dysphagia Symptom Questionnaire (DSQ) score ≥10 on a scale of 0 to 84. Patients were stratified based on age at the time of the screening visit (12 to 17 years of age vs. 18 years and older) and use of PPI at randomisation. TREET Part A was conducted first. TREET Part B opened after enrolment into TREET Part A was complete. Patients completing the 24 weeks of the double-blind treatment period in Parts A or B were provided an option to enrol in a 28-week active treatment extension study (TREET Part C).
In Part A, a total of 81 patients, of which 61 were adults and 20 were paediatric patients 12 to 17 years of age, were randomised to receive either 300 mg dupilumab every week (N=42) or placebo (N=39). In Part B, a total of 240 patients, of which 161 were adults and 79 were paediatric patients 12 to 17 years of age, were randomised to receive either 300 mg dupilumab every week (N=80), 300 mg dupilumab every other week (N=81; the 300 mg every other week dosage regimen is not approved for EoE) or placebo (N=79). In Part C, all patients who previously participated in Part A received 300 mg dupilumab (N=77) every week. Of the patients who previously participated in Part B, 111 received dupilumab 300 mg every week in Part C. Rescue with systemic and/or swallowed topical corticosteroids or emergency esophageal dilation was allowed during the study at the investigator's discretion.
In Part A, a total of 74.1% of patients enrolled had a history of prior use of swallowed topical corticosteroids for the treatment of EoE and 43.2% had a history of prior esophageal dilation. In Part B, a total of 73.3% of patients enrolled had a history of prior use of swallowed topical corticosteroids for the treatment of EoE and 35.4% had a history of prior esophageal dilation.
The co-primary efficacy endpoints in both trials were the proportion of patients achieving histological remission defined as peak esophageal intraepithelial eosinophil count of ≤6 eos/hpf at week 24 and the absolute change in the patient-reported DSQ score from baseline to week 24. Secondary endpoints included change from baseline in the following: percent change in peak esophageal intraepithelial eosinophil count (eos/hpf), absolute change in Mean Grade Score from the Histology Scoring System (EoEHSS), absolute change in Mean Stage Score from the EoEHSS, absolute change in EoEEndoscopic Reference Score (EoE-EREFS), and proportion of patients achieving peak esophageal intraepithelial eosinophil count of <15 eos/hpf.
The demographics and baseline characteristics of TREET Parts A and B are provided in Table 28.
Table 28. Demographics and baseline characteristics (TREET Parts A and B):
| Parameter | TREET Part Α (N=81) | TREET Part Β (N=240) |
|---|---|---|
| Age (years), mean (SD) | 31.5 (14.3) | 28.1 (13.1) |
| % Male | 60.5 | 63.8 |
| % White | 96.3 | 90.4 |
| Weight (kg), mean (SD) | 77.8 (21.0) | 76.2 (20.6) |
| BMI (kg/m2), mean (SD) | 26.1 (6.3) | 25.7 (6.2) |
| Duration of EoE (yr), mean (SD) | 5.01 (4.3) | 5.57 (4.8) |
| Prior swallowed topical steroid use (%) | 74.1 | 73.3 |
| Prior esophageal dilations (%) | 43.2 | 35.4 |
| PPI use at randomisation (%) | 67.9 | 72.5 |
| Food elimination diet at screening (%) | 40.7 | 37.1 |
| DSQ (0-84a), mean (SD) | 33.6 (12.4) | 36.7 (11.2) |
| Peak esophageal intraepithelial EOS count of 3 regions, mean (SD) | 89.3 (48.3) | 87.1 (45.8) |
| Mean esophageal intraepithelial EOS count of 3 regions, mean (SD) | 64.3 (37.6) | 60.5 (32.9) |
| EoEHSS grade Score [0-3a], mean (SD) | 1.3 (0.4) | 1.3 (0.4) |
| EoEHSS stage Score [0-3a], mean (SD) | 1.3 (0.4) | 1.3 (0.3) |
| EREFS total Score [0-18a], mean (SD) | 6.3 (2.8) | 7.2 (3.2) |
a Higher scores indicate greater disease severity
SD = standard deviation
The results for TREET Parts A and B are presented in Table 29.
Table 29. Efficacy results of dupilumab at week 24 in patients 12 years of age and older with EoE (TREET Parts A and B):
| TREET Part A | TREET Part B | |||||
|---|---|---|---|---|---|---|
| Dupilumab 300 mg QW N=42 | Placebo N=39 | Difference vs. placebo (95% CI)d | Dupilumab 300 mg QW N=80 | Placebo N=79 | Difference vs. placebo (95% CI)d | |
| Co-primary endpoints | ||||||
| Proportion of patients achieving histological remission (peak esophageal intraepithelial eosinophil count ≤6 eos/hpf), n (%) | 25 (59.5) | 2 (5.1) | 55.3 (39.58, 71.04) | 47 (58.8) | 5 (6.3) | 53.5 (41.20, 65.79) |
| Absolute change from baseline in DSQ score (0-84a), LS mean (SE) | -21.92 (2.53) | -9.60 (2.79) | -12.32 (-19.11, -5.54) | -23.78 (1.86) | -13.86 (1.91) | -9.92 (-14.81, -5.02) |
| Secondary endpoints | ||||||
| Percent change from baseline in peak esophageal intraepithelial eosinophil count, LS mean (SE) | -71.24 (6.95) | -2.98 (7.60) | -68.26 (-86.90, -49.62) | -80.24 (8.34) | 8.38 (10.09) | -88.62 (-112.19, 65.05) |
| Absolute change from baseline in EoEHSS mean grade score (0-3b), LS mean (SE) | -0.76 (0.06) | -0.00 (0.06) | -0.76 (-0.91, -0.61) | -0.83 (0.04) | -0.15 (0.05) | -0.682 (-0.79, -0.57) |
| Absolute change from baseline in EoEHSS mean stage score (0-3b), LS mean (SE) | -0.75 (0.06) | -0.01 (0.06) | -0.74 (-0.88, -0.60) | -0.80 (0.04) | -0.13 (0.04) | -0.672 (-0.78, -0.57) |
| Absolute change from baseline in EoE-EREFS (0-18c), LS mean (SE) | -3.2 (0.41) | -0.3 (0.41) | -2.9 (-3.91, -1.84) | -4.5 (0.36) | -0.6 (0.38) | -3.8 (-4.77, -2.93) |
| Proportion of patients achieving peak esophageal intraepithelial eosinophil count of <15 eos/hpf, n (%) | 27 (64.3) | 3 (7.7) | 57 (41.69, 73.33) | 66 (82.5) | 6 (7.6) | 74.9 (64.25, 85.5) |
a Total biweekly DSQ scores range from 0 to 84; higher scores indicate greater frequency and severity of dysphagia
b EoEHSS scores range from 0 to 3; higher scores indicate greater severity and extent of histological abnormalities
c EoE-EREFS overall scores range from 0 to 18; higher scores indicate worse endoscopic inflammatory and remodeling findings
d LS mean difference for continuous endpoints and absolute difference in proportions for categorical endpoints
The efficacy results for co-primary and key secondary endpoints in prior swallowed topical corticosteroids subgroup and in patients who were inadequately controlled, intolerant or contraindicated to swallowed topical corticosteroids were consistent with the overall population.
In Parts A and B, a greater proportion of patients randomised to dupilumab achieved histological remission (peak esophageal intraepithelial eosinophil count ≤6 eos/hpf) compared to placebo. The proportion of patients with histological remission observed after 24 weeks of treatment in Part A and B was maintained for 52 weeks in Part C. Similarly, other histological and endoscopic improvement were maintained through 52 weeks.
Treatment with dupilumab also resulted in a significant improvement in LS mean change in DSQ score compared to placebo as early as week 4 and were maintained through week 24. Efficacy in part C was similar to results observed in Parts A and B, with a continuous improvement for DSQ up to 52 weeks (TREET Part A and C Figure 9 and TREET Parts B and C Figure 10).
Figure 9. LS mean change from baseline in DSQ score over time in patients 12 years of age and older with EoE (TREET Part A and C):

Figure 10. Mean change from baseline in DSQ score over time in patients 12 years of age and older with EoE (TREET Parts B and C):

Consistent with improvement in DSQ total score in TREET Parts A and B, nominally significant improvements were observed at week 24 compared to placebo in pain related to dysphagia (DSQ pain score), health-related QoL (EoE-IQ), and the frequency of other non-dysphagia symptoms (EoE-SQ).
Paediatric Patients 1 to 11 Years of Age with EoE
The efficacy and safety of dupilumab was evaluated in paediatric patients 1 to 11 years of age with EoE in a two-part study up to 52-weeks (EoE KIDS Part A & Part B). All enrolled patients had to have failed conventional medicinal therapy (proton pump inhibitors), 77.5% were treated with another conventional medicinal therapy (swallowed topical corticosteroids) prior to inclusion, and 53.5% of patients were inadequately controlled, intolerant or contraindicated to swallowed topical corticosteroid treatment. Eligible patients had ≥15 intraepithelial eosinophils per high-power field (eos/hpf) despite a treatment course of a proton pump inhibitor (PPI) either prior to or during the screening period and a history of EoE signs and symptoms. Part A was a 16-week randomized, double-blind, parallel-group, multicenter, placebo-controlled trial. Part B was an active treatment extension period evaluating the dupilumab regimens for an additional 36 weeks.
Part A evaluated dupilumab versus matching placebo at dosing regimens based on body weight (≥5 to <15 kg (100 mg Q2W), ≥15 to <30 kg (200 mg Q2W) and ≥30 to <60 kg (300 mg Q2W). The recommended dosing regimen of dupilumab was selected for paediatric patients 1 to 11 years of age weighing ≥40 kg (300 mg QW) based upon simulations with a population pharmacokinetic model to match exposures of adult and paediatric patients 12 to 17 years of age with EoE receiving 300 mg QW for whom histologic and symptomatic efficacy were observed [see section 5.1 and section 5.2].
A total of 71 patients were enrolled in Part A. The mean age was 7 years (range 1 to 11 years), the median weight was 24.8 kg, 74.6% of patients were male, 87.3% were White, 9.9% were Black, and 1.4% were Asian. A total of 55 patients from Part A continued in Part B.
The primary efficacy endpoint in Part A was the proportion of patients achieving histological remission defined as peak esophageal intraepithelial eosinophil count of ≤6 eos/hpf at Week 16. Secondary endpoints included the proportion of patients achieving peak esophageal intraepithelial eosinophil count of <15 eos/hpf and the change from baseline in the following: peak esophageal intraepithelial eosinophil count (eos/hpf), absolute change in Mean Grade Score from the Histology Scoring System (EoEHSS), absolute change in Mean Stage Score from the EoEHSS, and absolute change in EoE-Endoscopic Reference Score (EoE-EREFS). The impact on signs of EoE was measured using observer reported outcomes; Paediatric EoE Sign/Symptom Questionnaire-Caregiver (PESQ-C) assessed the proportion of days with one or more EoE signs and Paediatric Eosinophilic Esophagitis Symptom Score (PEESS) assessed the frequency and severity of EoE signs.
Efficacy results for Part A are presented in Table 30 and below.
Table 30. Efficacy Results of dupilumab at Week 16 in Subjects 1 to 11 Years of Age with EoE (EoE KIDS Part A):
| Dupilumaba N=37 | Placebo N=34 | Difference vs Placebo (95% CI) | |
|---|---|---|---|
| Primary Endpoint | |||
| Proportion of subjects achieving histological remission (peak esophageal intraepithelial eosinophil count ≤6 eos/hpf), n (%)b | 25 (67.6) | 1 (2.9) | 64.5 (48.19, 80.85) |
| Secondary Endpoints | |||
| Proportion of subjects achieving peak esophageal intraepithelial eosinophil count of <15 eos/hpf, n (%)b | 31 (83.8) | 1 (2.9) | 81 (68.07, 94.10) |
| Percent change from baseline in peak esophageal intraepithelial eosinophil count (eos/hpf), LS mean (SE)c | -86.09 (11.84) | 20.98 (12.23) | -107.07 (-139.25, -74.90) |
| Absolute change in Mean Grade Score (0-3d) from the Histology Scoring System (EoEHSS) from baseline, LS mean (SE) | -0.879 (0.05) | 0.023 (0.05) | -0.902 (-1.03, -0.77) |
| Absolute change in Mean Stage Score (0-3d) from the EoEHSS from baseline, LS mean (SE) | -0.835 (0.05) | 0.048 (0.05) | -0.883 (-1.01, -0.76) |
| Absolute change in EoE- Endoscopic Reference Score(EoE-EREFS) (0-18e) from baseline, LS mean (SE) | -3.5 (0.42) | 0.3 (0.45) | -3.8 (-4.94, -2.63) |
a DUPIXENT was evaluated at tiered dosing regimens based on body weight: ≥5 to <15 kg (100 mg Q2W), ≥15 to <30 kg (200 mg Q2W), and ≥30 to <60 kg (300 mg Q2W).
b For histological remission, the difference in percentages is estimated using the Mantel-Haenszel method, adjusting for baseline weight group (≥5 to <15 kg, ≥15 to <30 kg, and ≥30 to <60 kg).
c The difference in absolute change or percent change is estimated using ANCOVA model with baseline measurement as covariate and the treatment, baseline weight group (≥5 to <15 kg, ≥15 to <30 kg, and ≥30 to <60 kg) strata as fixed factors.
d EoEHSS scores range from 0 to 3; higher scores indicate greater severity and extent of histological abnormalities.
e EoE-EREFS overall scores range from 0 to 18; higher scores indicate worse end oscopic inflammatory and remodelling findings.
In Part A, a greater proportion of patients randomized to dupilumab achieved histological remission (peak esophageal intraepithelial eosinophil count ≤6 eos/hpf) compared to placebo. The proportion of subjects with histological remission observed after 16 weeks of treatment in Part A was maintained for 52 weeks in Part B.
Numerical improvement in the proportion of days with 1 or more EoE signs (PESQ-C) was observed after 16 weeks of dupilumab treatment in Part A and was maintained for 52 weeks in Part B.
Nominally significant improvement in the frequency and severity of EoE signs (PEESS-Caregiver) was observed after 16 weeks of treatment in Part A. PEESS-Caregiver was not measured in Part B.
Clinical efficacy in chronic obstructive pulmonary disease (COPD)
The chronic obstructive pulmonary disease (COPD) program included two randomized, double-blind, multicenter, parallel-group, placebo-controlled trials (BOREAS and NOTUS) of 52 weeks in treatment duration which enrolled a total of 1874 adult patients with COPD to evaluate dupilumab as add-on maintenance therapy.
Both trials enrolled patients with a diagnosis of COPD with moderate to severe airflow limitation (post-bronchodilator FEV1/FVC ratio <0.7 and post-bronchodilator FEV1 of 30% to 70% predicted), chronic productive cough for at least 3 months in the past year, and a minimum blood eosinophil count of 300 cells/mcL at screening. Patients were uncontrolled with a Medical Research Council (MRC) dyspnoea score ≥2 (range 0-4) and an exacerbation history of at least 2 moderate or 1 severe exacerbation in the previous year despite receiving maintenance triple therapy consisting of a long-acting muscarinic antagonist (LAMA), long-acting beta agonist (LABA), and inhaled corticosteroid (ICS). Patients were allowed to receive maintenance therapy consisting of a LAMA and LABA if an ICS was not appropriate. Exacerbations were defined as moderate severity if treatment with systemic corticosteroids and/or antibiotics was required or severe if they resulted in hospitalization or observation for over 24 hours in an emergency department or urgent care facility.
In both trials, patients were randomized to receive dupilumab 300 mg every two weeks (Q2W) or placebo in addition to their background maintenance therapy for 52 weeks.
In both trials, the primary endpoint was the annualized rate of moderate or severe COPD exacerbations during the 52-week treatment period. Secondary endpoints included change from baseline in pre-bronchodilator FEV1 in the overall population and in the subgroup of patients with baseline FeNO ≥20 ppb at Weeks 12 and 52, change from baseline in St. George's Respiratory Questionnaire (SGRQ) total score at Week 52, and annualized rate of moderate or severe COPD exacerbations in the subgroup of patients with baseline FeNO ≥20 ppb during the 52-week treatment period.
The demographics and baseline characteristics of BOREAS and NOTUS are provided in Table 28.
Table 28. Demographics and baseline characteristics (BOREAS and NOTUS):
| Parameter | BOREAS (N=939) | NOTUS (N=935) |
|---|---|---|
| Mean age (years) (± SD) | 65.1 (8.1) | 65.0 (8.3) |
| Male (%) | 66.0 | 67.6 |
| White (%)c | 84.1 | 89.6 |
| Mean smoking history (pack-years) (± SD) | 40.48 (23.35) | 40.3 (27.2) |
| Current Smokers (%) | 30 | 29.5 |
| Emphysema (%) | 32.6 | 30.4 |
| Mean Duration of COPD (years) (± SD) | 8.8 (6.0) | 9.3 (6.4) |
| Mean number of moderatea or severeb exacerbations in previous year (± SD) | 2.3 (1.0) | 2.1 (0.9) |
| Mean number of severe exacerbationsb in previous year (± SD) | 0.3 (0.7) | 0.3 (0.6) |
| Background COPD medications at randomization: ICS/LAMA/LABA (%) | 97.6 | 98.8 |
| Mean post-bronchodilator FEV1/FVC ratio (± SD) | 0.49 (0.12) | 0.50 (0.12) |
| Mean pre-bronchodilator FEV1 (L) (± SD) | 1.30 (0.46) | 1.36 (0.50) |
| Mean post-bronchodilator FEV1 (L) (± SD) | 1.40 (0.47) | 1.45 (0.49) |
| Mean percent predicted post-bronchodilator FEV1 (%) (± SD) | 50.6 (13.1) | 50.1 (12.6) |
| Mean percent predicted post-bronchodilator FEV1 <50% (%) (± SD) | 467 (49.7) | 478 (51.3) |
| Mean SGRQ Total score (± SD) | 48.42 (17.42) | 51.5 (17.0) |
| Mean E-RS:COPD [total score] (± SD) | 12.9 (7.1) | 13.3 (7.0) |
| Mean BODE index score (± SD) | 4.06 (1.66) | 4.0 (1.6) |
| Mean FeNO ppb (± SD) | 24.3 (22.4) | 24.6 (26.0) |
| Mean baseline blood eosinophil count (cells/mcL) (± SD) | 401 (298) | 407 (336) |
| Median baseline blood eosinophil count (cells/mcL) (Q1-Q3) | 340 (240-460) | 330 (220-460) |
ICS = inhaled corticosteroid; LAMA = long acting muscarinic antagonist; LABA = long acting beta agonist, FEV1= forced expiratory volume in 1 second; FVC = forced vital capacity; FeNO = fraction of exhaled nitric oxide; BODE = body-mass index, airflow obstruction, dyspnea, exercise capacity
a Exacerbations treated with either systemic corticosteroids and/or antibiotics
b Exacerbations requiring hospitalization or observation for over 24 hours in an emergency department or urgent care facility
c In BOREAS, 0.5% of participants were Black and 14.3% were Asian. In NOTUS, 1.3% of participants were Black and 1.1% were Asian
Exacerbations
In both trials, dupilumab demonstrated a statistically significant reduction in the annualized rate of moderate or severe COPD exacerbations compared to placebo when added to background maintenance therapy (see Table 29).
Table 29. Annualized Rate of moderatea or severeb COPD exacerbations in BOREAS and NOTUS:
| Trial | Treatment (N) | Rate (exacerbations/year) | Rate Ratio vs. Placebo (95% CI) | % Reduction in Exacerbation Rate vs. Placebo |
|---|---|---|---|---|
| Primary Endpoint: Moderatea or Severeb COPD exacerbations | ||||
| BOREAS | Dupilumab 300 mg Q2W (N=468) | 0.78 | 0.705 (0.581, 0.857)c | 30% |
| Placebo (N=471) | 1.10 | |||
| NOTUS | Dupilumab 300 mg Q2W (N=470) | 0.86 | 0.664 (0.535, 0.823)d | 34% |
| Placebo (N=465) | 1.30 | |||
| Pooled Component of Primary Endpointe: Severeb COPD exacerbations | ||||
| BOREAS and NOTUS | Dupilumab 300 mg Q2W (N=938) | 0.08 | 0.674 (0.438 to 1.037) | 33% |
| Placebo (N=936) | 0.12 | |||
a Exacerbations treated with either systemic corticosteroids and/or antibiotics
b Exacerbations requiring hospitalization, or observation for >24 hours in an emergency department/urgent care facility or resulting in death
c p value = 0.0005
d p value = 0.0002
e Analysis of the component of the primary endpoint was not adjusted for multiplicity
In both trials, the cumulative mean number of moderate or severe exacerbations observed over 52 weeks was lower in patients receiving dupilumab compared to placebo (see Figure 11a and 11b).
Figure 11. Cumulative mean number of moderate or severe COPD exacerbations over 52 weeks in BOREAS and NOTUS:
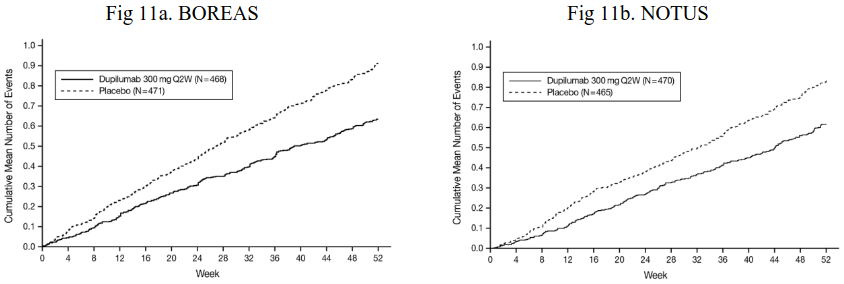
The time to first moderate or severe COPD exacerbation was longer for patients receiving dupilumab compared to placebo in BOREAS (HR: 0.803; 95% CI: 0.658, 0.980) and NOTUS (HR: 0.71.; 95% CI: 0.57, 0.889).
In the subgroup analysis of patients with a higher baseline FeNO (≥ 20 ppb) in BOREAS (N=383), treatment with dupilumab statistically significantly reduced the annualized rate of moderate or severe COPD exacerbations compared to placebo (Rate Ratio: 0.625; 95% CI: 0.45, 0.869; p=0.005). In NOTUS, treatment with dupilumab showed a nominally significant reduction in the annualized rate of moderate or severe COPD exacerbations in the subgroup of patients with a higher baseline FeNO (≥20 ppb) (N=355) compared to placebo (Rate Ratio: 0.471; 95% CI: 0.328, 0.675; p<0.0001).
Reductions in the annualized rate of moderate or severe exacerbations were observed across all predefined subgroups including age, gender, race, smoking status, blood eosinophil counts, number of exacerbations in previous year (≤2, 3, and ≥4), high-dose ICS at baseline, and baseline percent predicted post-bronchodilator FEV1 (<50%, ≥50%). In patients with emphysema, reduction in the rate of moderate or severe exacerbations was consistent with the overall population.
Lung Function
In both trials, dupilumab demonstrated a statistically significant improvement in pre-bronchodilator FEV1 at Weeks 12 and 52 compared to placebo when added to background maintenance therapy (see Table 30). Greater improvements in lung function (LS mean change from baseline in pre-bronchodilator FEV1) were observed in patients treated with dupilumab compared to placebo as early as Week 2 (BOREAS) (first assessment) and Week 4 (NOTUS) and were sustained at Week 52 (see Figures 12a and 12b).
In BOREAS, rapid improvements in post-bronchodilator FEV1, post-bronchodilator FEV1/FVC ratio, and pre-bronchodilator FVC were observed with dupilumab treatment compared to placebo as early as Week 2 (first assessment) and were maintained through Week 52. In NOTUS, rapid improvements in post-bronchodilator FEV1 and post-bronchodilator FEV1/FVC ratio were observed with dupilumab treatment compared to placebo as early as week 8 and week 2, respectively, and were maintained through Week 52.
Table 30. Mean change from baseline for lung function endpoints in BOREAS and NOTUS:
| BOREAS | NOTUS | |||||
|---|---|---|---|---|---|---|
| Dupilumab (N=468) | Placebo (N=471) | Difference (95% CI) for Dupilumab vs. Placebo | Dupilumab (N=470) | Placebo (N=465) | Difference (95% CI) for Dupilumab vs. Placebo | |
| Change from baseline in pre- bronchodilator FEV1 at Week 12, LS Mean (SE) | 0.160 (0.018) | 0.077 (0.018) | 0.083 (0.042 to 0.125)a | 0.139 (0.017) | 0.057 (0.017) | 0.082 (0.040 to 0.124)f |
| Change from baseline in pre- bronchodilator FEV1 at Week 52, LS Mean (SE)k | 0.153 (0.019) | 0.070 (0.019) | 0.083 (0.038 to 0.128)b | 0.115 (0.021) | 0.054 (0.020) | 0.062 (0.011 to 0.113)g |
| Change from baseline in post- bronchodilator FEV1 at Week 12, LS Mean (SE) | 0.156 (0.018) | 0.084 (0.018) | 0.072 (0.030 to 0.115)c | 0.136 (0.020) | 0.064 (0.020) | 0.072 (0.023 to 0.121)h |
| Change from baseline in post- bronchodilator FEV1/FVC ratio at Week 12, LS Mean (SE) | 0.037 (0.004) | 0.023 (0.004) | 0.014 (0.005 to 0.023)d | 0.030 (0.004) | 0.013 (0.004) | 0.017 (0.006 to 0.028)i |
| Change from baseline in pre- bronchodilator FVC at Week 12, LS Mean (SE) | 0.098 (0.022) | 0.029 (0.022) | 0.069 (0.016 to 0.121)e | 0.083 (0.024) | 0.018 (0.024) | 0.066 (0.005 to 0.126)j |
LS = least squares, SE = standard error, FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity
a p-value < 0.0001, bp-value = 0.0003 (all statistically significant vs placebo with adjustment for multiplicity)
cnominal p-value = 0.0010, dnominal p-value = 0.0016 enominal p-value = 0.0103
f p-value=0.0001, gp-value=0.0182 (all statistically significant vs placebo with adjustment for multiplicity)
h nominal p-value=0.0042, inominal p-value=0.0020, jnominal p-value=0.0327
k Efficacy results for mean change from baseline in pre-bronchodilator FEV1 at Week 52 are presented for 721 out of 935 patients who completed the 52-week treatment period or had discontinued the trial at the time of data analysis.
Figure 12. Mean change from baseline in pre-bronchodilator FEV1 (L) over time in BOREAS and NOTUSa:
a Efficacy results for mean change from baseline in pre-bronchodilator FEV1 over time are presented for 721 out of 935 patients who completed the 52-week treatment period or had discontinued the trial at the time of data analysis.
In the subgroup analysis of patients with higher baseline FeNO (≥20 ppb) in BOREAS (N=383), treatment with dupilumab statistically significantly improved pre-bronchodilator FEV1 from baseline at Week 12 (LS mean change: 0.232 dupilumab vs 0.108 placebo; LS mean difference: 0.124 [95% CI: 0.045, 0.203]; p=0.002) and Week 52 (LS mean change: 0.247 dupilumab vs 0.120 placebo; LS mean difference: 0.127 [95% CI: 0.042, 0.212]; p=0.003) compared to placebo. In NOTUS, statistically significant improvement from baseline was observed in the subgroup of patients with a higher baseline FeNO (≥20 ppb) at Week 12 (N=355; LS mean change: 0.221 dupilumab vs 0.081 placebo; LS mean difference: 0.141 [95% CI: 0.058, 0.223]; p=0.001). Treatment with dupilumab improved pre-50 bronchodilator FEV1 at Week 52 in the subgroup of patients with higher baseline FeNO (≥20 ppb) compared to placebo in NOTUS (N=264; LS mean change: 0.176 dupilumab vs 0.095 placebo; LS mean difference: 0.081[95% CI: -0.019, 0.181]) but did not meet statistical significance.
Improvements in lung function as measured by pre-bronchodilator FEV1 were observed across all predefined subgroups including age, gender, race, smoking status, blood eosinophil counts, number of exacerbations in previous year (≤2, 3, and ≥4), high-dose ICS at baseline, and baseline percent predicted post-bronchodilator FEV1 (<50%, ≥50%). In patients with emphysema, improvement in lung function as measured by pre-bronchodilator FEV1 was consistent with the overall population.
Health-Related Quality of Life
In BOREAS, a statistically significant improvement in SGRQ total score was observed in patients treated with dupilumab compared to placebo (LS mean change: -9.73 dupilumab vs -6.37 placebo; LS mean difference: -3.36 [95% CI: -5.46, -1.27]; p=0.0017). In NOTUS, dupilumab nominally improved SGRQ total score at Week 52 compared to placebo (LS mean change: -9.82 dupilumab vs -6.44 placebo; LS mean difference: -3.37; 95% CI: -5.81, -0.93]; p=0.007).
Patients with post-bronchodilator FEV1 <30% or >70%
Patients with post-bronchodilator FEV1 <30% or >70% at screening were excluded from BOREAS and NOTUS. However, limited data are available in patients with post-bronchodilator FEV1 <30% or >70% at baseline.
Paediatric population
Atopic dermatitis
The safety and efficacy of dupilumab have been established in paediatric patients 6 months of age and older with atopic dermatitis. Use of dupilumab in this age group is supported by study AD-1526 which included 251 adolescents aged 12 to 17 years old with moderate-to-severe atopic dermatitis, in study AD-1652 which included 367 paediatric patients aged 6 to 11 years old with severe atopic dermatitis, and study AD-1539 which included 162 children ages 6 months to 5 years old with moderate-to-severe atopic dermatitis (125 of whom had severe atopic dermatitis). Long term use is supported by study AD-1434 which enrolled 823 paediatric patients aged 6 months to 17 years of age, this included 275 adolescents, 368 children 6 to 11 years of age, and 180 children 6 months to 5 years of age. The safety and efficacy were generally consistent between children 6 months to 5 years old, 6 to 11 years old, adolescent (12 to 17 years old), and adult patients with atopic dermatitis (see section 4.8). Safety and efficacy in paediatric patients <6 months of age with atopic dermatitis have not been established.
Asthma
A total of 107 adolescents aged 12 to 17 years with moderate to severe asthma were enrolled in QUEST study and received either 200 mg (N=21) or 300 mg (N=18) dupilumab (or matching placebo either 200 mg [N=34] or 300 mg [N=34]) every other week. Efficacy with respect to severe asthma exacerbations and lung function was observed in both adolescents and adults. For both the 200 mg and 300 mg every other week doses, significant improvements in FEV1 (LS mean change from baseline at week 12) were observed (0.36 L and 0.27 L, respectively). For the 200 mg every other week dose, patients had a reduction in the rate of severe exacerbations that was consistent with adults. The safety profile in adolescents was generally similar to the adults.
A total of 89 adolescents aged 12 to 17 years with moderate-to-severe asthma were enrolled in the open label long-term study (TRAVERSE). In this study, efficacy was measured as a secondary endpoint, was similar to results observed in the pivotal studies and was sustained up to 96 weeks.
A total of 408 children aged 6 to 11 years with moderate-to-severe asthma was enrolled in the VOYAGE study, which evaluated doses of 100 mg Q2W and 200 mg Q2W. The efficacy of dupilumab 300 mg Q4W in children aged 6 to 11 years is extrapolated from the efficacy of 100 mg and 200 mg Q2W in VOYAGE and 200 mg and 300 mg Q2W in adults and adolescents (QUEST). Patients who completed the treatment period of the VOYAGE study could participate in the open label extension study (EXCURSION). Eighteen patients (≥15 kg to <30 kg) out of 365 patients were exposed to 300 mg Q4W in this study, and the safety profile was similar to that seen in VOYAGE. Safety and efficacy in paediatric patients <6 years of age with asthma have not been established.
Eosinophilic Esophagitis
The safety and efficacy of dupilumab for the treatment of EoE have been established in paediatric patients 1 to 17 years of age. Use of dupilumab in this population is supported by adequate and well-controlled studies and additional pharmacokinetic data. A total of 72 paediatric patients 12 to 17 years of age received dupilumab 300 mg QW or placebo for 24 weeks (TREET Parts A and B). Of these, there were 37 dupilumab treated patients in Parts A and B; 34 continued treatment with 300 mg QW for an additional 28 weeks (TREET Part C). A total of 71 paediatric patients 1 to 11 years of age received dupilumab 100 mg Q2W, 200 mg Q2W, 300 mg Q2W, or placebo for 16 weeks (EoE KIDS Part A). Of these, there were 37 dupilumab treated patients in Part A all of whom continued treatment with these dupilumab regimens for an additional 36 weeks (EoE KIDS Part B). The use of dupilumab 300 mg QW in patients 1 to 11 years of age with EoE with a body weight ≥40 kg is also supported by a population pharmacokinetic analysis [see section 5.1]. The safety and efficacy of dupilumab in adults and paediatric patients were similar [see section 4.8 and section 5.1].
Chronic Spontaneous Urticaria
A total of 12 adolescents aged 12 to 17 years with CSU were enrolled in CUPID Study A, B, and C and received dupilumab 200 mg Q2W (30 kg to <60 kg), 300 mg Q2W (≥60 kg) or placebo. The effectiveness of dupilumab for the treatment of CSU in adolescents is based on extrapolation of efficacy in adults with this condition. The recommended dosage in adolescents is based on body weight.
The European Medicines Agency has deferred the obligation to submit the results of studies with dupilumab in one or more subset of the paediatric population in asthma (see section 4.2 for information on paediatric use). The European Medicines Agency has waived the obligation to submit the results of studies with dupilumab in all subsets of the paediatric population in the treatment of nasal polyposis, prurigo nodularis and chronic obstructive pulmonary disease (see section 4.2 for information on paediatric use). Obligations related to the paediatric investigation plans for atopic dermatitis and EoE have been fulfilled.
Pharmacokinetic properties
The pharmacokinetics of dupilumab is similar in patients with atopic dermatitis, asthma, CRSwNP, PN, EoE, and COPD.
Absorption
After a single subcutaneous (SC) dose of 75-600 mg dupilumab to adults, median times to maximum concentration in serum (tmax) were 3-7 days. The absolute bioavailability of dupilumab following a SC dose is similar between AD, asthma, CRSwNP, EoE, COPD, and CSU patients, ranging between 61% and 64%, as determined by a population pharmacokinetics (PK) analysis.
Steady-state concentrations were achieved by week 16 following the administration of 600 mg starting dose and 300 mg dose every other week or 300 mg dose every other week without a loading dose. Across clinical trials, the mean ±SD steady-state trough concentrations ranged from 55.3±34.3 mcg/mL to 81.5±43.9 mcg/mL for 300 mg administered Q2W, from 172±76.6 mcg/ml to 195±71.7 mcg/ml for 300 mg administered weekly, and from 29.2±18.7 to 36.5±22.2 mcg/mL for 200 mg administered Q2W.
Distribution
A volume of distribution for dupilumab of approximately 4.6 L was estimated by population PK analysis, indicating that dupilumab is distributed primarily in the vascular system.
Biotransformation
Specific metabolism studies were not conducted because dupilumab is a protein. Dupilumab is expected to degrade to small peptides and individual amino acids.
Elimination
Dupilumab elimination is mediated by parallel linear and nonlinear pathways. At higher concentrations, dupilumab elimination is primarily through a non-saturable proteolytic pathway, while at lower concentrations, the non-linear saturable IL-4R α target-mediated elimination predominates. After the last steady state dose of 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W, or 200 mg Q4W dupilumab, the median times to decrease below the lower limit of detection, estimated by population PK analysis, ranged from 9-13 weeks in adults and adolescents and are approximately 1.5 times and 2.5 times longer in paediatric patients 6 to 11 years of age and paediatric patients less than 6 years of age, respectively.
Linearity/non-linearity
Due to nonlinear clearance, dupilumab exposure, as measured by area under the concentration-time curve, increases with dose in a greater than proportional manner following single SC doses from 75-600 mg.
Special populations
Gender
Gender was not found to be associated with any clinically meaningful impact on the systemic exposure of dupilumab determined by population PK analysis.
Elderly
Of the 1539 patients with atopic dermatitis, including patients with atopic hand and foot dermatitis exposed to dupilumab in a phase 2 dose-ranging study or phase 3 placebo-controlled studies, a total of 71 were 65 years or older. Although no differences in safety or efficacy were observed between older and younger adult atopic dermatitis patients, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients.
Age was not found to be associated with any clinically meaningful impact on the systemic exposure of dupilumab determined by population PK analysis. However, there were only 61 patients over 65 years of age included in this analysis.
Of the 1,977 patients with asthma exposed to dupilumab, a total of 240 patients were 65 years or older and 39 patients were 75 years or older. Efficacy and safety in this age group were similar to the overall study population.
There were only 79 patients older than 65 years with CRSwNP exposed to dupilumab among them 11 patients were 75 years and older.
Of the 152 patients with PN exposed to dupilumab, a total of 37 were 65 years of age or older. A total of 8 patients were 75 years of age or older. Efficacy and safety in these age groups were similar to the overall study population.
There were only 2 patients older than 65 years with EoE exposed to dupilumab.
Of the 938 patients with COPD exposed to dupilumab, a total of 551 were 65 years of age and older including 116 patients 75 years of age and older. Efficacy and safety in this age group were similar to the overall study population.
Of the 198 patients with CSU exposed to dupilumab, a total of 30 were 65 years of age and older including 7 patients 75 years of age and older. Efficacy and safety in this age group were similar to the overall study population.
Race
Race was not found to be associated with any clinically meaningful impact on the systemic exposure of dupilumab by population PK analysis.
Hepatic impairment
Dupilumab, as a monoclonal antibody, is not expected to undergo significant hepatic elimination. No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of dupilumab.
Renal impairment
Dupilumab, as a monoclonal antibody, is not expected to undergo significant renal elimination. No clinical studies have been conducted to evaluate the effect of renal impairment on the pharmacokinetics of dupilumab. Population PK analysis did not identify mild or moderate renal impairment as having a clinically meaningful influence on the systemic exposure of dupilumab. Very limited data are available in patients with severe renal impairment.
Body weight
Dupilumab trough concentrations were lower in subjects with higher body weight with no meaningful impact on efficacy. There were only 6 patients exposed to dupilumab with body weight ≥130 kg in CRSwNP clinical studies.
Paediatric population
Atopic dermatitis:
Based on population pharmacokinetic analysis, age did not affect dupilumab clearance in adults and in paediatric patients 6 to 17 years of age. In paediatric patients from 6 months to 5 years of age, clearance increased with age but is accommodated in the recommended dose regimen.
The pharmacokinetics of dupilumab in paediatric patients (<6 months of age) or body weight <5 kg with atopic dermatitis has not been studied.
For adolescents 12 to 17 years of age with atopic dermatitis receiving every other week dosing (Q2W) with either 200 mg (<60 kg) or 300 mg (≥60 kg), the mean ± SD steady state trough concentration of dupilumab was 54.5 ± 27.0 mcg/mL.
For children 6 to 11 years of age with atopic dermatitis receiving every four week dosing (Q4W) with 300 mg (≥15 kg) in AD-1652, the mean ± SD steady-state trough concentration was 76.3 ± 37.2 mcg/mL. At week 16 in AD-1434 in children 6 to 11 years of age who initiated every four week dosing (Q4W) with 300 mg (≥15 kg), and whose dose was increased to every other week dosing (Q2W) with 200 mg (≥15 to <60 kg) or 300 mg (≥60 kg), the mean ± SD steady-state trough concentration was 108 ± 53.8 mcg/mL. For children 6 to 11 years of age receiving 300 mg Q4W, initial doses of 300 mg on Days 1 and 15 produce similar steady-state exposure as an initial dose of 600 mg on Day 1, based on PK simulations.
For children 6 months to 5 years of age with atopic dermatitis receiving every four week dosing (Q4W) with 300 mg (≥15 to <30 kg) or 200 mg (≥5 to <15 kg) mean ± SD steady-state trough concentration was 110 ± 42.8 mcg/mL and 109 ± 50.8 mcg/mL, respectively.
Asthma:
The pharmacokinetics of dupilumab in paediatric patients (<6 years of age) with asthma has not been studied.
A total of 107 adolescents aged 12 to 17 years with asthma were enrolled in QUEST study. The mean ± SD steady-state trough concentrations of dupilumab were 107 ± 51.6 mcg/mL and 46.7 ± 26.9 mcg/mL, respectively, for 300 mg or 200 mg administered every other week. No age-related pharmacokinetic difference was observed in adolescent patients after correction for body weight.
In the VOYAGE study, dupilumab pharmacokinetics was investigated in 270 patients with moderate-to-severe asthma following subcutaneous administration of either 100 mg Q2W (for 91 children weighing <30 kg) or 200 mg Q2W (for 179 children weighing ≥30 kg). The volume of distribution for dupilumab of approximately 3.7 L was estimated by population PK analysis. Steady-state concentrations were achieved by week 12. The mean ± SD steady-state trough concentration was 58.4 ± 28.0 mcg/mL and 85.1 ± 44.9 mcg/mL, respectively. Simulation of a 300 mg Q4W subcutaneous dose in children aged 6 to 11 years with body weight of ≥15 kg to <30 kg and ≥30 kg to <60 kg resulted in predicted steady-state trough concentrations similar to the observed trough concentrations of 200 mg Q2W (≥30 kg) and 100 mg Q2W (<30 kg), respectively. In addition, simulation of a 300 mg Q4W subcutaneous dose in children aged 6 to 11 years with body weight of ≥15 kg to <60 kg resulted in predicted steady-state trough concentrations similar to those demonstrated to be efficacious in adults and adolescents. After the last steady state dose, the median time for dupilumab concentrations to decrease below the lower limit of detection, estimated by population PK analysis, was 14 to 18 weeks for 100 mg Q2W, 200 mg Q2W or 300 mg Q4W.
CRSwNP:
CRSwNP does not normally occur in children. The pharmacokinetics of dupilumab in paediatric patients (<18 years of age) with CRSwNP has not been studied.
PN:
The pharmacokinetics of dupilumab in paediatric patients (< 18 years of age) with PN has not been studied.
Eosinophilic esophagitis:
A total of 35 adolescents aged 12 to 17 years with eosinophilic esophagitis weighing ≥40 kg were enrolled in TREET Parts A and B, receiving 300 mg every week dosing (QW). The mean ± SD steady-state trough concentration of dupilumab was 227 ± 95.3 mcg/mL.
In a clinical study (EoE KIDS Part A), dupilumab pharmacokinetics were investigated in 36 children 1 to 11 years of age with EoE receiving dupilumab [≥5 to <15 kg (100 mg Q2W), ≥15 to <30 kg (200 mg Q2W), and ≥30 to <60 kg (300 mg Q2W)], the mean ± SD steady-state trough concentration of dupilumab was 163±60.8 mcg/mL.
Simulations for paediatric patients 1 to 11 years of age were conducted with a population pharmacokinetic model to predict trough concentrations of dupilumab at steady-state as follows: ≥15 to <30 kg receiving 200 mg Q2W (170±78 mcg/mL); ≥30 to <40 kg receiving 300 mg Q2W (158±63 mcg/mL); or ≥40 kg receiving 300 mg QW (276±99 mcg/mL). Steady-state trough concentrations were also simulated for adult and paediatric patients 12 to 17 years of age and patients from ≥30 to <40 kg receiving 300 mg Q2W (159±61 mcg/mL).
COPD:
COPD does not normally occur in children. The pharmacokinetics of dupilumab have not been studied in paediatric patients (<18 years of age) with COPD.
Chronic Spontaneous Urticaria:
Pharmacokinetics in paediatric patients (<12 years of age) with CSU have not been established.
A total of 12 adolescents aged 12 to 17 years with CSU were enrolled in CUPID Study A, B, and C. The observed steady-state trough concentrations of the 5 adolescent patients with CSU who received dupilumab 300 mg Q2W or 200 mg Q2W for 24 weeks were within the range of the individual steady-state trough concentrations in adult patients with CSU who received dupilumab 300 mg Q2W for 24 weeks.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity (including safety pharmacology endpoints) and toxicity to reproduction and development.
The mutagenic potential of dupilumab has not been evaluated; however monoclonal antibodies are not expected to alter DNA or chromosomes.
Carcinogenicity studies have not been conducted with dupilumab. An evaluation of the available evidence related to IL-4Rα inhibition and animal toxicology data with surrogate antibodies does not suggest an increased carcinogenic potential for dupilumab.
During a reproductive toxicology study conducted in monkeys, using a surrogate antibody specific to the monkey IL-4Rα, no foetal abnormalities were observed at doses that saturate the IL-4Rα.
An enhanced pre- and post-natal developmental study revealed no adverse effects in maternal animals or their offspring up to 6 months post-partum/post-birth.
Fertility studies conducted in male and female mice using a surrogate antibody against IL-4Rα showed no impairment of fertility (see section 4.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.