EGRIFTA SV Solution for injection Ref.[108542] Active ingredients: Tesamorelin
Source: FDA, National Drug Code (US) Revision Year: 2023
12.1. Mechanism of Action
In vitro, tesamorelin binds and stimulates human GRF receptors with similar potency as the endogenous GRF [see Clinical Pharmacology (12.2)].
Growth hormone-releasing factor (GHRF), also known as growth hormone-releasing hormone (GHRH), is a hypothalamic peptide that acts on the pituitary somatotroph cells to stimulate the synthesis and pulsatile release of endogenous growth hormone (GH), which is both anabolic and lipolytic. GH exerts its effects by interacting with specific receptors on a variety of target cells, including chondrocytes, osteoblasts, myocytes, hepatocytes, and adipocytes, resulting in a host of pharmacodynamic effects. Some, but not all these effects, are primarily mediated by IGF-1 produced in the liver and in peripheral tissues.
12.2. Pharmacodynamics
Tesamorelin stimulates growth hormone secretion, and subsequently increases IGF-1 and IGFBP-3 levels. No clinically significant changes in the levels of other pituitary hormones, including thyroid-stimulating hormone (TSH), luteinizing hormone (LH), adrenocorticotropic hormone (ACTH) and prolactin, were observed in patients receiving EGRIFTA in clinical trials.
12.3. Pharmacokinetics
Absorption
The absolute bioavailability of tesamorelin after subcutaneous administration of a 2 mg dose of EGRIFTA (1 mg/vial formulation) was determined to be less than 4% in healthy adult subjects.
Single and multiple dose pharmacokinetics have been characterized in healthy subjects and HIV-infected patients without lipodystrophy using a 2 mg dose of EGRIFTA (1 mg/vial formulation). Tesamorelin mean extent of absorption (AUC) was 34% higher in HIV-infected patients than healthy subjects. Tesamorelin peak plasma concentration (Cmax) was similar in HIV-infected patients and healthy subjects. The median peak plasma tesamorelin concentration (Tmax) was 0.15 h in both populations.
Following single dose of subcutaneous administration of 1.4 mg of EGRIFTA SV (2 mg/vial formulation) in healthy subjects, the mean [coefficient of variation (CV)] AUC0-inf was 889.1 (57%) pg.h/mL. The mean (CV) Cmax value was 2956.1 (47%) pg/mL and the median Tmax was 0.15 h.
The systemic exposure (Cmax and AUCs) of tesamorelin is similar between the 1.4 mg dose of EGRIFTA SV (2 mg/vial formulation) and the 2 mg dose of EGRIFTA (1 mg/vial formulation).
Distribution
The mean volume of distribution (±SD) of tesamorelin following a single subcutaneous administration of the 1.4 mg dose of EGRIFTA SV (2 mg/vial formulation) was 4.8 ± 1.9 L/kg in healthy subjects.
Metabolism
No formal metabolism studies have been performed in humans.
Elimination
Mean elimination half-life (t1/2) of tesamorelin was 8 minutes in healthy subjects after single dose subcutaneous administration of the 1.4 mg of EGRIFTA SV (2 mg/vial formulation).
Specific Populations
Pharmacokinetics of tesamorelin in patients with renal or hepatic impairment, in pediatric patients, or in elderly patients has not been established.
Drug Interactions
Simvastatin
The effect of multiple dose administration of EGRIFTA on the pharmacokinetics of simvastatin and simvastatin acid was evaluated in healthy subjects. Co-administration with simvastatin (a CYP3A substrate) resulted in 8% decrease in extent of absorption (AUCinf) and 5% increase in rate of absorption (Cmax) of simvastatin. For simvastatin acid there was a 15% decrease in AUCinf and 1% decrease in Cmax [see Drug Interactions (7.1)].
Ritonavir
The effect of multiple dose administration of EGRIFTA on the pharmacokinetics of ritonavir was evaluated in healthy subjects. Co-administration with ritonavir resulted in 9% decrease in AUCinf and 11% decrease in Cmax of ritonavir [see Drug Interactions (7.1)].
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Life-time carcinogenicity studies in rodents have not been conducted with tesamorelin acetate. No potential mutagenicity of tesamorelin acetate was revealed in a battery of tests including induction of gene mutations in bacteria (the Ames test), gene mutations in mammalian cells grown in vitro (hamster CHOK1 cells), and chromosomal damage in intact animals (bone marrow cells in mice). There was no effect on fertility in male or female rats following administration of tesamorelin acetate at doses up to 0.6 mg/kg (approximately equal to clinical exposure) for 28 days in males or 14 days in females.
14. Clinical Studies
The safety and effectiveness of EGRIFTA SV (2 mg/vial formulation) has been established based on adequate and well controlled studies with EGRIFTA (1 mg/vial formulation), as well as a demonstration of comparable bioavailability between the 1.4 mg EGRIFTA SV dose (2 mg/vial formulation) and the 2 mg EGRIFTA dose (1 mg/vial formulation) [see Clinical Pharmacology (12.3)].
Two multicenter, randomized, double-blind, placebo-controlled studies were conducted in HIV-infected patients with lipodystrophy and excess abdominal fat (abdominal lipohypertrophy). Study 1 and Study 2 consisted of a 26-week Main Phase and a 26-week Extension Phase, respectively. Main inclusion criteria were age 18 to 65 years, a waist circumference ≥95 cm (37.4 inches) and a waist-to-hip ratio ≥0.94 for men and ≥94 cm (37.0 inches) and ≥0.88 for women, respectively, and fasting blood glucose (FBG) <150 mg/dL (8.33 mmol/L). Main exclusion criteria included BMI ≤20 kg/m², type 1 diabetes mellitus, type 2 diabetes mellitus, previous treatment with insulin or with oral hypoglycemic or insulin-sensitizing agents, history of malignancy, and hypopituitarism. Patients were on a stable anti-retroviral regimen for at least 8 weeks prior to randomization. Patients meeting the inclusion/exclusion criteria were randomized in a 2:1 ratio to receive a 2 mg dose of EGRIFTA (1 mg/vial formulation) or placebo subcutaneously daily for 26 weeks. The primary efficacy assessment for each of these studies was the percent change from baseline to Week 26 in visceral adipose tissue (VAT), as assessed by computed tomography (CT) scan at L4-L5 vertebral level. Secondary endpoints included changes from baseline in patient-reported outcomes related to body image, triglycerides, ratio of total cholesterol to HDL cholesterol, IGF-1 levels, and safety parameters. Other endpoints included changes from baseline in waist circumference, abdominal subcutaneous tissue (SAT), trunk fat, and lean body mass. In both studies, EGRIFTA-treated patients completing the 26-week treatment period were re-randomized to blinded therapy with either daily placebo or a 2 mg dose of EGRIFTA (1 mg/vial formulation) for an additional 26-week treatment period (Extension Phase) in order to assess maintenance of VAT reduction and to gather long-term safety data. For inclusion in the Extension Phase studies, subjects must have completed the Main Phase with FBG ≤150 mg/dL.
Main Phase (Baseline to Week 26)
Study 1 (NCT 00123253)
This study randomized 412 HIV-infected patients with lipodystrophy and excess abdominal fat to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=273) or placebo (N=137). At baseline for the two groups combined, mean age was 48 years; 86% were male; 75% were white, 14% were Black/African American, and 8% were Hispanic; mean weight was 90 kg; mean BMI was 29 kg/m²; mean waist circumference was 104 cm; mean hip circumference was 100 cm; mean VAT was 176 cm²; mean CD4 cell count was 606 cells/mm³; 69% had undetectable viral load (<50 copies/mL); and 33.7% randomized to EGRIFTA and 36.6% randomized to placebo had impaired glucose tolerance, while 5.6% randomized to EGRIFTA and 6.7% randomized to placebo had diet-controlled diabetes mellitus. The twenty-six week completion rate in Study 1 was 80%.
Study 2 (NCT 00435136)
This study randomized 404 HIV-infected patients with lipodystrophy and excess abdominal fat to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=270) or placebo (N=126). At baseline for the two groups combined, mean age was 48 years; 84% were male; 77% were white, 12% were Black/African American, and 9% were Hispanic; mean weight was 88 kg; mean BMI was 29 kg/m²; mean waist circumference was 105 cm; mean hip circumference was 100 cm; mean VAT was 189 cm²; mean CD4 cell count was 592 cells/mm³; 83% had undetectable viral load (<50 copies/mL); and 44% randomized to EGRIFTA and 40% randomized to placebo had impaired glucose tolerance, while 9% randomized to EGRIFTA and 10% randomized to placebo had diet-controlled type 2 diabetes mellitus. The twenty-six week completion rate in Study 2 was 74%.
Results for the Main Phases of Studies 1 and 2 are presented in Tables 2 and 3.
Table 2. Changes from Baseline to Week 26 in Visceral Adipose Tissue (cm²) by Treatment Group (Intent-To-Treat Population with Last Observation Carried Forward):
| MAIN PHASE (Baseline-Week 26) | |||||
|---|---|---|---|---|---|
| Study 1 | Study 2 | ||||
| 2 mg EGRIFTA (1 mg/vial) (N=273) | Placebo (N=137) | 2 mg EGRIFTA (1 mg/vial) (N=270) | Placebo (N=126) | ||
| Baseline (cm²) | 178 (77) | 171 (77) | 186 (87) | 195 (95) | |
| Change (cm²) | -27 | 4 | -21 | -0 | |
| Mean treatment difference (95% CI) | -31 (-39,-24) | -21 (-29,-12) | |||
| Mean change (%)1 | -18 | 2 | -14 | -2 | |
| Mean treatment difference (95% CI)1 | -20 (-24, -15) | -12 (-16, -7) | |||
Baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval.
1 Results derived from the statistical model: Ln(VAT Week 26/VAT Baseline) = Ln(VAT Baseline) + treatment group
Table 3. Changes from Baseline to Week 26 in IGF-1, IGFBP-3, Weight, and Waist Circumference by Treatment Group (Intent-To-Treat Population with Last Observation Carried Forward):
| MAIN PHASE (Baseline-Week 26) | |||||
|---|---|---|---|---|---|
| Study 1 | Study 2 | ||||
| 2 mg EGRIFTA (1 mg/vial) (N=273) | Placebo (N=137) | 2 mg EGRIFTA (1 mg/vial) (N=270) | Placebo (N=126) | ||
| IGF-1 (ng/mL) | Baseline | 161 (59) | 168 (75) | 146 (66) | 149 (59) |
| Change | 107 | -15 | 108 | 3 | |
| Mean treatment difference (95% CI) | 122 (101, 141) | 105 (85, 126) | |||
| IGFBP-3 (mg/L) | Baseline | 3 (1) | 3 (1) | 3 (1) | 3 (1) |
| Change | 0.4 | -0.2 | 0.8 | -0.0 | |
| Mean treatment difference (95% CI) | 0.6 (0.5, 0.8) | 0.8 (0.5, 1.0) | |||
| Weight (kg) | Baseline | 90 (14) | 90 (14) | 89 (14) | 87 (16) |
| Change | -0.4 | 0.0 | 0.5 | 0.3 | |
| Mean treatment difference (95% CI) | -0.4 (-1.3, 0.5) | 0.2 (-0.7, 1.3) | |||
| Waist circumference (cm) | Baseline | 104 (10) | 105 (9) | 105 (9) | 105 (9) |
| Change | -3 (5) | -1 (4) | -2 (5) | -1 (5) | |
| Mean treatment difference (95% CI) | -2 (-2.8, -0.9) | -1 (-2.5, -0.3) | |||
Baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval.
At Week 26, treatment with a 2 mg dose of EGRIFTA (1 mg/vial formulation) resulted in a reduction from baseline in mean trunk fat of 1.0 kg in Study 1 and 0.8 kg in Study 2, respectively (compared with an increase of 0.4 kg in Study 1 and of 0.2 kg in Study 2, respectively, in patients receiving placebo). Treatment with EGRIFTA resulted in an increase from baseline in mean lean body mass of 1.3 kg in Study 1 and of 1.2 kg in Study 2, respectively (compared with a decrease of 0.2 kg in Study 1 and of 0.03 kg in Study 2, respectively, in patients receiving placebo).
Patient Reported Outcomes
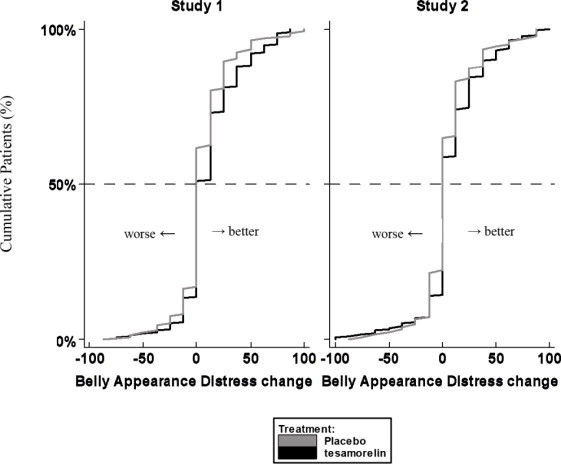
Patients rated the degree of distress associated with their belly appearance on a 9-point rating scale that was then transformed to a score from 0 (extremely upsetting and distressing) to 100 (extremely encouraging). A score of 50 indicated neutral (no feeling either way). A positive change from baseline score indicated improvement, i.e., less distress.
The cumulative distribution of response (change from baseline to 26 weeks) is shown in Figure 1 for both treatment groups. A curve shifted to the right on this scale indicates a greater percentage of patients reporting improvement.
Figure 1. Cumulative Distribution of Response for Belly Appearance Distress:
Extension Phase (Weeks 26-52)
In the double-blind Extension Phase, patients on a 2 mg dose of EGRIFTA (1 mg/vial formulation) completing the 26-week Main Phase were re-randomized to receive a 2 mg dose of EGRIFTA (1 mg/vial formulation) or placebo.
Study 1 (NCT 00123253)
This study re-randomized 207 HIV-infected patients with lipodystrophy who completed a 2 mg dose of EGRIFTA (1 mg/vial formulation) treatment in the Main Phase to receive either EGRIFTA (N=154) or placebo (N=50) for an additional 26-week duration (3:1 randomization ratio). At baseline (Week 26) for the two groups combined, mean age was 48 years; 88% were male; 78% were white, 12% were Black/African American, and 8% were Hispanic; mean weight was 90 kg; mean BMI was 29 kg/m²; mean waist circumference was 102 cm; mean hip circumference was 100 cm; mean VAT was 145 cm²; mean CD4 cell count was 639 cells/mm³; 68% had undetectable viral load (<50 copies/mL); and for those EGRIFTA-treated patients completing the 26-week treatment period that were re-randomized to EGRIFTA (T-T group) or re-randomized to placebo, 37% and 32%, respectively, had impaired glucose tolerance, while 2% re-randomized to EGRIFTA and 6% re-randomized to placebo had diet-controlled type 2 diabetes mellitus. The completion rate for patients randomized into the extension phase of Study 1 was 83%.
Study 2 (NCT 00435136)
This study re-randomized 177 HIV-infected patients with lipodystrophy who completed EGRIFTA treatment in the Main Phase to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=92) or placebo (N=85) for an additional 26-week duration (1:1 randomization ratio). At baseline (Week 26) for the two groups combined, mean age was 48 years; 90% were male; 84% were white, 9% were Black/African American, and 7% were Hispanic; mean weight was 89 kg; mean BMI was 28 kg/m²; mean waist circumference was 105 cm; mean hip circumference was 100 cm; mean VAT was 172 cm²; mean CD4 cell count was 579 cells/mm³; 82% had undetectable viral load (<50 copies/mL); and for those EGRIFTA-treated patients completing the 26-week treatment period that were re-randomized to EGRIFTA (T-T group) or re-randomized to placebo, 49% and 51%, respectively, had impaired glucose tolerance, while 4% re-randomized to EGRIFTA and 13% re-randomized to placebo had diet-controlled diabetes mellitus. The completion rate for patients randomized into the extension phase of Study 2 was 81%.
Results for the Extension Phases of Studies 1 and 2 are presented in Tables 4 and 5.
Table 4. Changes from Week 26 Baseline to Week 52 in Visceral Adipose Tissue (cm²) by Treatment Group (Intent-To-Treat Population with Last Observation Carried Forward):
| EXTENSION PHASE (Week 26-52) | ||||
|---|---|---|---|---|
| Study 1 | Study 2 | |||
| T-T1 (Week 26-52) (N=154) | T-P2 (Week 26-52) (N=50) | T-T1 (Week 26-52) (N=92) | T-P2 (Week 26-52) (N=85) | |
| Week 26 (cm²) | 145 (72) | 144 (72) | 166 (89) | 177 (88) |
| Change (cm²) | 3 | 25 | -11 | 24 |
| Mean treatment difference (95% CI) | -22 (-34, -10) | -35 (-48, -22) | ||
| Mean change (%)3 | 0 | 22 | -5 | 16 |
| Mean treatment difference (95% CI)3 | -17 (-24, -10) | -18 (-24, -11) | ||
Week 26 baseline data are expressed as mean (SD). Change refers to least-squares mean (LSM); CI: confidence interval.
1 T-T = tesamorelin for Weeks 0-26 and tesamorelin for Weeks 26-52
2 T-P = tesamorelin for Weeks 0-26 and placebo for Weeks 26-52
3 Results derived from the statistical model: Ln(VAT Week 52/Week 26) = Ln(Week 26 VAT) + treatment group
Figure 2 shows the percent change in VAT from baseline (Week 0) over time until 52 weeks in completer patients.
Figure 2. Percent Change from Baseline in VAT over Time

Data in Figure 2 are expressed as mean values. T-T (tesamorelin to tesamorelin) refers to the group of patients who received tesamorelin for Weeks 0-26 and were re-randomized to tesamorelin for Weeks 26-52. T-P (tesamorelin to placebo) refers to the group of patients who received tesamorelin for Weeks 0-26 and were re-randomized to placebo for Weeks 26-52. P-T (placebo to tesamorelin) refers to the group of patients who received placebo for Weeks 0-26 and were switched to tesamorelin (treated open label) for Weeks 26-52.
Table 5. Changes from Week 26 Baseline to Week 52 in IGF-1, IGFBP-3, Weight, and Waist Circumference by Treatment Group (Intent-To-Treat Population with Last Observation Carried Forward):
| EXTENSION PHASE (Weeks 26-52) | |||||
|---|---|---|---|---|---|
| Study 1 | Study 2 | ||||
| T-T1 (Week 26-52) (N=154) | T-P2 (Week 26-52) (N=50) | T-T1 (Week 26-52) (N=92) | T-P2 (Week 26-52) (N=85) | ||
| IGF-1 (ng/mL) | Week 26 | 291 (124) | 281 (105) | 280 (134) | 269 (110) |
| Change | -59 | -137 | -25 | -135 | |
| Mean treatment difference (95% CI) | 78 (50, 106) | 110 (87, 134) | |||
| IGFBP-3 (mg/L) | Week 26 | 3 (1) | 3 (1) | 3 (1) | 3 (1) |
| Change | -0.2 | -0.5 | -0.3 | -0.9 | |
| Mean treatment difference (95% CI) | 0.3 (-0.0, 0.6) | 0.6 (0.3, 0.9) | |||
| Weight (kg) | Week 26 | 89 (14) | 92 (17) | 89 (13) | 90 (14) |
| Change | 0.2 | 0.6 | -0.5 | 0.1 | |
| Mean treatment difference (95% CI) | -0.4 (-2, 1) | -0.6 (-2, 1) | |||
| Waist circumference (cm) | Week 26 | 101 (10) | 102 (12) | 101 (9) | 103 (11) |
| Change | -0.2 | 2.4 | -1.1 | 0.2 | |
| Mean treatment difference (95% CI) | -2.6 (-4, -1) | -1.3 (-2, 0) | |||
Week 26 baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval.
1 T-T = tesamorelin for Week 0-26 and tesamorelin for Week 26-52
2 T-P = tesamorelin for Week 0-26 and placebo for Week 26-52
Patients treated with a 2 mg dose of EGRIFTA (1 mg/formulation) for 52 weeks (T-T group) showed no change between Weeks 26 and 52 in mean trunk fat (increase of 0.1 kg in Study 1 and decrease of 0.5 kg in Study 2, respectively, compared with an increase of 1.4 kg in patients in the T-P group in Study 1 and an increase of 1.09 kg in Study 2, respectively) nor was there a change from Week 26 baseline in mean lean body mass (decrease of 0.1 kg in Study 1 and increase of 0.1 kg in Study 2, respectively, compared with a decrease of 1.8 kg in patients in the T-P group in Study 1 and a decrease of 1.7 kg in Study 2, respectively).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.