EVRYSDI Powder for oral solution Ref.[27849] Active ingredients: Risdiplam
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other drugs for disorders of the musculo-skeletal system
ATC code: M09AX10
Mechanism of action
Risdiplam is a survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier designed to treat SMA caused by mutations of the SMN1 gene in chromosome 5q that lead to SMN protein deficiency. Functional SMN protein deficiency is directly linked to the SMA pathophysiology which includes progressive loss of motor neurons and muscle weakness. Risdiplam corrects the splicing of SMN2 to shift the balance from exon 7 exclusion to exon 7 inclusion into the mRNA transcript, leading to an increased production of functional and stable SMN protein. Thus, risdiplam treats SMA by increasing and sustaining functional SMN protein levels.
Pharmacodynamic effects
In the studies FIREFISH (patients aged 2-7 months at enrolment), SUNFISH (patients aged 2-25 years at enrolment), and JEWELFISH (patients aged 1-60 years at enrolment) in infantile-onset SMA and later-onset SMA patients, risdiplam led to an increase in SMN protein in blood with a greater than 2-fold median change from baseline within 4 weeks of treatment initiation across all SMA types studied. The increase was sustained throughout the treatment period (of at least 24 months).
Cardiac electrophysiology
The effect of risdiplam on the QTc interval was evaluated in a study in 47 healthy adult subjects. At the therapeutic exposure, risdiplam did not prolong the QTc interval.
Clinical efficacy and safety
The efficacy of Evrysdi for the treatment of SMA patients with infantile-onset (SMA Type 1) and later-onset SMA (SMA type 2 and 3) was evaluated in 2 pivotal clinical studies, FIREFISH and SUNFISH. Efficacy data of Evrysdi for the treatment of pre-symptomatic SMA patients was evaluated in the RAINBOWFISH clinical study. Patients with a clinical diagnosis of Type 4 SMA have not been studied in clinical trials.
Infantile-onset SMA
tudy BP39056 (FIREFISH) is an open-label, 2-part study to investigate the efficacy, safety, PK and pharmacodynamics (PD) of Evrysdi in symptomatic Type 1 SMA patients (all patients had genetically confirmed disease with 2 copies of the SMN2 gene). Part 1 of FIREFISH was designed as a dose-finding part of the study. The confirmatory Part 2 of the FIREFISH study assessed the efficacy of Evrysdi. Patients from Part 1 did not take part in Part 2.
The key efficacy endpoint was the ability to sit without support for at least 5 seconds, as measured by Item 22 of the Bayley Scales of Infant and Toddler Development – Third Edition (BSID-III) gross motor scale, after 12 months of treatment.
FIREFISH Part 2
In FIREFISH Part 2, 41 patients with Type 1 SMA were enrolled. The median age of onset of clinical signs and symptoms of Type 1 SMA was 1.5 months (range: 1.0-3.0 months), 54% were female, 54% Caucasian and 34% Asian. The median age at enrolment was 5.3 months (range: 2.2-6.9 months) and the median time between onset of symptoms and first dose was 3.4 months (range: 1.0-6.0 months). At baseline, the median Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disease (CHOP-INTEND) score was 22.0 points (range: 8.0-37.0) and the median Hammersmith Infant Neurological Examination Module 2 (HINE-2) score was 1.0 (range: 0.0-5.0).
The primary endpoint was the proportion of patients with the ability to sit without support for at least 5 seconds after 12 months of treatment (BSID-III gross motor scale, Item 22). The key efficacy endpoints of Evrysdi treated patients are shown in Table 3.
Table 3. Summary of key efficacy results at month 12 (FIREFISH Part 2):
| Efficacy Endpoints | Proportion of Patients N=41 (90% CI) | |
|---|---|---|
| Month 12 | Month 24 | |
| Motor function and development milestones | ||
| BSID-III: sitting without support for at least 5 seconds | 29.3% (17.8%, 43.1%) p<0.0001a | 61.0% (46.9%, 73.8%) |
| CHOP-INTEND: score of 40 or higher | 56.1% (42.1%, 69.4%) | 75.6% (62.2%, 86.1%) |
| CHOP-INTEND: increase of ≥4 points from baseline | 90.2% (79.1%, 96.6%) | 90.2% (79.1%, 96.6%) |
| HINE-2: motor milestone respondersb | 78.0% (64.8%, 88.0%) | 85.4% (73.2%, 93.4%) |
| HINE-2: sitting without supportc | 24.4% (13.9%, 37.9%) | 53.7% (39.8%, 67.1%) |
| Survival and event-free survival | ||
| Event-free survivald | 85.4% (73.4%, 92.2%) | 82.9% (70.5%, 90.4%) |
| Alive | 92.7% (82.2%, 97.1%) | 92.7% (82.2%, 97.1%) |
| Feeding | ||
| Ability to feed orallye | 82.9% (70.3%, 91.7%) | 85.4% (73.2%, 93.4%) |
Abbreviations: CHOP-INTEND=Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HINE2=Module 2 of the Hammersmith Infant Neurological Examination.
a p-value is based on a one-sided exact binomial test. The result is compared to a threshold of 5%.
b According to HINE-2: ≥2 point increase [or maximal score] in ability to kick, OR ≥1 point increase in the motor milestones of head control, rolling, sitting, crawling, standing or walking, AND improvement in more categories of motor milestones than worsening is defined as a responder for this analysis.
c Sitting without support includes patients that achieved “stable sit” (24%, 10/41) and “pivots (rotates)” (29%, 12/41) as assessed by the HINE-2 at Month 24.
d An event is meeting the endpoint of permanent ventilation defined as tracheostomy or ≥16 hours of non-invasive ventilation per day or intubation for >21 consecutive days in the absence of, or following the resolution of, an acute reversible event. Three patients died within the first 3 months following study enrolment and 4 patients met the endpoint of permanent ventilation before Month 24. These 4 patients achieved an increase of at least 4 points in their CHOPINTEND score from baseline.
e Includes patients who were fed exclusively orally (29 patients overall) and those who were fed orally in combination with a feeding tube (6 patients overall) at Month 24.
At Month 24, 44% of patients achieved sitting without support for 30 seconds (BSID-III, Item 26). Patients continued to achieve additional motor milestones as measured by the HINE-2: 80.5% were able to roll, and 27% of patients achieved a standing measure (12% supporting weight and 15% standing with support).
Untreated patients with infantile-onset SMA would never be able to sit without support and only 25% would be expected to survive without permanent ventilation beyond 14 months of age.
Figure 1. Kaplan-Meier plot of event-free survival (FIREFISH Part 1 and Part 2):

+ Censored: two patients in Part 2 were censored because the patients attended the Month 24 visit early, one patient in Part 1 was censored after discontinuing treatment and died 3.5 months later
Figure 2. Mean change from baseline in CHOP-INTEND total score (FIREFISH Part 2):

FIREFISH Part 1
The efficacy of Evrysdi in Type 1 SMA patients is also supported by results from FIREFISH Part 1. For the 21 patients from Part 1, the baseline characteristics were consistent with symptomatic patients with Type 1 SMA. The median age at enrollment was 6.7 months (range: 3.3-6.9 months) and the median time between onset of symptoms and first dose was 4.0 months (range: 2.0-5.8 months).
A total of 17 patients received the therapeutic dose of Evrysdi (dose selected for Part 2). After 12 months of treatment, 41% (7/17) of these patients were able to sit independently for at least 5 seconds (BSID-III, Item 22). After 24 months of treatment, 3 more patients receiving the therapeutic dose were able to sit independently for at least 5 seconds, leading to a total of 10 patients (59%) achieving this motor milestone.
After 12 months of treatment, 90% (19/21) of patients were alive and event-free (without permanent ventilation) and reached 15 months of age or older. After a minimum of 33 months of treatment, 81% (17/21) of patients were alive and event-free and reached an age of 37 months or older (median 41
months; range 37 to 53 months), see Figure 1. Three patients died during treatment and one patient died 3.5 months after discontinuing treatment.
Later Onset SMA
Study BP39055 (SUNFISH), is a 2-part, multicentre study to investigate the efficacy, safety, PK and PD of Evrysdi in SMA Type 2 or Type 3 patients between 2-25 years of age. Part 1 was the exploratory dose-finding portion and Part 2 was the randomized, double-blind, placebo-controlled confirmatory portion. Patients from Part 1 did not take part in Part 2.
The primary endpoint was the change from baseline score at Month 12 on the Motor Function Measure-32 (MFM32). The MFM32 has the ability to assess a wide range of motor function across a broad range of SMA patients. The total MFM32 score is expressed as a percentage (range: 0-100) of the maximum possible score, with higher scores indicating greater motor function.
SUNFISH Part 2
SUNFISH Part 2 is the randomized, double-blinded, placebo-controlled portion of the SUNFISH study in 180 non-ambulant patients with Type 2 (71%) or Type 3 (29%) SMA. Patients were randomized with 2:1 ratio to receive either Evrysdi at the therapeutic dose (see section 4.2) or placebo. Randomization was stratified by age group (2 to 5, 6 to 11, 12 to 17, 18 to 25 years old).
The median age of patients at the start of treatment was 9.0 years old (range 2-25 years old), the median time between onset of initial SMA symptoms to first treatment was 102.6 (1-275) months. Overall, 30% were 2 to 5 years of age, 32% were 6 to 11 years of age, 26% were 12-17 years of age, and 12% were 18 to 25 years of age at study enrolment. Of the 180 patients included in the study, 51% were female, 67% Caucasian and 19% Asian. At baseline, 67% of patients had scoliosis (32% of patients with severe scoliosis). Patients had a mean baseline MFM32 score of 46.1 and Revised Upper Limb Module (RULM) score of 20.1. The baseline demographic characteristics were balanced between Evrysdi and placebo arms with the exception of scoliosis (63% of patients in the Evrysdi arm and 73% of patients in the placebo control).
The primary analysis for SUNFISH Part 2, the change from baseline in MFM32 total score at Month 12, showed a clinically meaningful and statistically significant difference between patients treated with Evrysdi and placebo. The results of the primary analysis and key secondary endpoints are shown in Table 4, Figure 3, and Figure 4.
Table 4. Summary of efficacy in patients with later-onset SMA at month 12 of treatment (SUNFISH Part 2):
| Endpoint | Evrysdi (N=120) | Placebo (N=60) |
|---|---|---|
| Primary Endpoint | ||
| Change from baseline in MFM32 total score1 at Month 12 LS mean (95%, CI) | 1.36 (0.61, 2.11) | -0.19 (-1.22, 0.84) |
| Difference from placebo Estimate (95% CI) p-value2 | 1.55 (0.30, 2.81) 0.0156 | |
| Secondary Endpoints | ||
| Proportion of patients with a change from baseline in MFM32 total score1 of 3 or more at Month 12 (95% CI)1 | 38.3% (28.9, 47.6) | 23.7% (12.0, 35.4) |
| Odds ratio for overall response (95% CI) Adjusted(unadjusted) p-value3,4 | 2.35 (1.01, 5.44) 0.0469 (0.0469) | |
| Change from baseline in RULM total score5 at Month 12 LS mean (95% CI) | 1.61 (1.00, 2.22) | 0.02 (-0.83, 0.87) |
| Difference from placebo estimate (95% CI) Adjusted (unadjusted) p-value2,4 | 1.59 (0.55, 2.62) 0.0469 (0.0028) | |
LS=least squares
1 Based on the missing data rule for MFM32, 6 patients were excluded from the analysis (Evrysdi n=115; placebo control n=59).
2 Data analysed using a mixed model repeated measure with baseline total score, treatment, visit, age group, treatment-by-visit and baseline-by-visit.
3 Data analysed using logistic regression with baseline total score, treatment and age group.
4 The adjusted p-value was obtained for the endpoints included in the hierarchical testing and was derived based on all the p-values from endpoints in order of the hierarchy up to the current endpoint.
5 Based on the missing data rule for RULM, 3 patients were excluded from the analysis (Evrysdi n=119; placebo control n=58).
Upon completion of 12 months of treatment, 117 patients continued to receive Evrysdi. At the time of the 24 month analysis, these patients who were treated with Evrysdi for 24 months overall experienced maintenance of improvement in motor function between month 12 and month 24. The mean change from baseline for MFM32 was 1.83 (95% CI: 0.74, 2.92) and for RULM was 2.79 (95% CI: 1.94, 3.64).
Figure 3. Mean change from baseline in MFM32 total score over 12 months in SUNFISH Part 21:

1 The least squares (LS) mean difference for change from baseline in MFM32 score [95% CI].
Figure 4. Mean change from baseline in RULM total score over 12 months in SUNFISH Part 21:

1 The least squares (LS) mean difference for change from baseline in RULM score [95% CI].
SUNFISH Part 1
Efficacy in later-onset SMA patients was also supported by results from Part 1, the dose-finding part of SUNFISH. In Part 1, 51 patients with Type 2 and 3 SMA (including 7 ambulatory patients) between 2 to 25 years of age were enrolled. After 1 year of treatment there was a clinically meaningful improvement in motor function as measured by MFM32, with a mean change from baseline of 2.7 points (95% CI: 1.5, 3.8). The improvement in MFM32 was maintained up to 2 years on treatment (mean change of 2.7 points [95% CI: 1.2, 4.2]).
Use in patients previously treated with other SMA-modifying therapies (JEWELFISH)
Study BP39054 (JEWELFISH, n=174) is a single arm, open-label study to investigate the safety, tolerability, PK and PD of Evrysdi in patients with infantile-onset and later-onset SMA (median age 14 years [range 1-60 years]), who had previously received treatment with other approved (nusinersen n=76, onasemnogene abeparvovec n=14) or investigational SMA modifying therapies. At baseline, out of the 168 patients aged 2-60 years, 83% of patients had scoliosis and 63% had a Hammersmith Functional Motor Scale Expanded (HFMSE) score <10 points.
At the analysis at month 24 of treatment, patients 2-60 years of age showed overall stabilization in motor function in MFM-32 and RULM (n=137 and n=133, respectively). Patients less than 2 years (n=6) maintained or gained motor milestones such as head control, rolling and sitting independently. All ambulatory patients (aged 5-46 years, n=15) retained their ability to walk.
Pre-symptomatic SMA (RAINBOWFISH)
Study BN40703 (RAINBOWFISH) is an open-label, single-arm, multicenter clinical study to investigate the efficacy, safety, pharmacokinetics, and pharmacodynamics of Evrysdi in infants from birth to 6 weeks of age (at first dose) who have been genetically diagnosed with SMA but do not yet present with symptoms.
The efficacy in pre-symptomatic SMA patients was evaluated at Month 12 in 26 patients [intent-to-treat (ITT) population] treated with Evrysdi: eight patients, 13 patients, and 5 patients had 2, 3, and ≥4 copies of the SMN2 gene, respectively. The median age of these patients at first dose was 25 days (range: 16 to 41 days), 62% were female, and 85% were Caucasian. At baseline, the median CHOP-INTEND score was 51.5 (range: 35.0 to 62.0), the median HINE-2 score was 2.5 (range: 0 to 6.0), and the median ulnar nerve compound muscle action potential (CMAP) amplitude was 3.6 mV (range: 0.5 to 6.7 mV).
The primary efficacy population (N=5) included patients with 2 SMN2 copies and a baseline CMAP amplitude ≥1.5 mV. In these patients, the median CHOP-INTEND score was 48.0 (range: 36.0 to 52.0), the median HINE-2 score was 2.0 (range 1.0 to 3.0), and the median CMAP amplitude was 2.6 mV (range: 1.6 to 3.8 mV) at baseline.
The primary endpoint was the proportion of patients in the primary efficacy population with the ability to sit without support for at least 5 seconds (BSID-III gross motor scale, Item 22) at Month 12; a statistically significant and clinically meaningful proportion of patients achieved this milestone compared to the predefined performance criterion of 5%.
The key efficacy endpoints of Evrysdi treated patients are shown in Table 5 and 6, and in Figure 5.
Table 5. Sitting ability as defined by BSID-III Item 22 for pre-symptomatic patients at Month 12:
| Efficacy Endpoint | Population | ||
|---|---|---|---|
| Primary Efficacy (N=5) | Patients with 2 SMN2 copiesa (N=8) | ITT (N=26) | |
| Proportion of patients sitting without support for at least 5 seconds (BSID-III, Item 22); (90% CI) | 80% (34.3%, 99.0%) p<0.0001b | 87.5% (52.9%, 99.4%) | 96.2% (83.0%, 99.8%) |
Abbreviations: BSID-III = Bayley Scales of Infant and Toddler Development – Third Edition; CI=Confidence Interval; ITT=Intent-to-treat.
a Patients with 2 SMN2 copies had a median CMAP amplitude of 2.0 (range 0.5 – 3.8) at baseline.
b p-value is based on a one-sided exact binomial test. The result is compared to a threshold of 5%.
Additionally, 80% (4/5) of the primary efficacy population, 87.5% (7/8) of patients with 2 SMN2 copies, and 80.8% (21/26) of patients in the ITT population achieved sitting without support for 30 seconds (BSID-III, Item 26).
Patients in the ITT population also achieved motor milestones as measured by the HINE-2 at Month 12 (N=25). In this population 96.0% of patients could sit [1 patient (1/8 patients with 2 SMN2 copies) achieved stable sit and 23 patients (6/8, 13/13, 4/4 of patients with 2, 3, and ≥4 SMN2 copies, respectively) could pivot/rotate]. In addition, 84% of patients could stand; 32% (N=8) patients could stand with support (3/8, 3/13 and 2/4 patients with 2, 3, and ≥4 SMN2 copies, respectively) and 52% (N=13) patients could stand unaided (1/8, 10/13 and 2/4 of patients with 2, 3, and ≥4 SMN2 copies, respectively). Furthermore, 72% of patients could bounce, cruise or walk; 8% (N=2) patients could bounce (2/8 patients with 2 SMN2 copies), 16% (N=4) could cruise (3/13 and ¼ patients with 3 and ≥4 SMN2 copies, respectively) and 48% (N=12) could walk independently (1/8, 9/13 and 2/4 patients with 2, 3, and ≥4 SMN2 copies, respectively). Seven patients were not tested for walking at Month 12.
Table 6. Summary of key efficacy endpoints for pre-symptomatic patients at Month 12:
| Efficacy Endpoints | ITT population (N=26) |
|---|---|
| Motor Function | |
| Proportion of patients who achieve a Total score of 50 or higher in the CHOP-INTEND (90 CI%) | 92%a (76.9%, 98.6%) |
| Proportion of patients who achieve a Total score of 60 or higher in the CHOP-INTEND (90 CI%) | 80%a (62.5%, 91.8%) |
| Feeding | |
| Proportion of patients with the ability to feed orally (90 CI%) | 96.2%b (83.0%, 99.8%) |
| Healthcare Utilization | |
| Proportion of patients with no hospitalisationsc (90 CI%) | 92.3% (77.7%, 98.6%) |
| Event-Free Survivald | |
| Proportion of patients with Event-Free Survival (90 CI%) | 100% (100%, 100%) |
Abbreviations: CHOP-INTEND=Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CI=Confidence Interval
a Based on N=25
b One patient was not assessed.
c Hospitalisations include all hospital admissions which spanned at least two days, and which are not due to study requirements.
d An event refers to death or permanent ventilation; permanent ventilation is defined as tracheostomy or ≥16 hours of non-invasive ventilation per day or intubation for >21 consecutive days in the absence of, or following the resolution of, an acute reversible event.
Figure 5. Median Total CHOP-INTEND Scores by Visit and SMN2 copy number (ITT population):
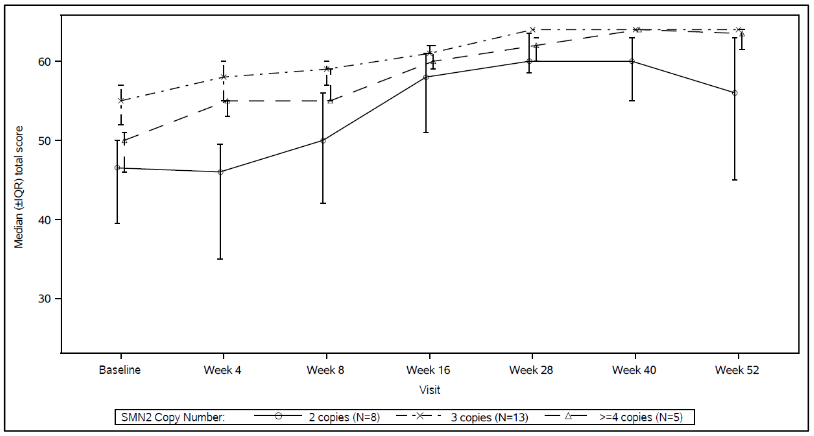
Abbreviations: IQR = Interquartile range; SMN2 = Survival of Motor Neuron 2.
5.2. Pharmacokinetic properties
Pharmacokinetic parameters have been characterised in healthy adult subjects and in patients with SMA.
After administration of treatment as an oral solution, PK of risdiplam were approximately linear between 0.6 and 18 mg. Risdiplam’s PK was best described by a population PK model with three-transit-compartment absorption, two-compartment disposition and first-order elimination. Body weight and age were found to have significant effect on the PK.
The estimated exposure (mean AUC0-24h) for infantile-onset SMA patients (age 2-7 months at enrolment) at the therapeutic dose of 0.2 mg/kg once daily was 1930 ng.h/mL. The estimated mean exposure in pre-symptomatic infants (16 days to <2 months of age) in the RAINBOWFISH study was 2020 ng.h/mL at 0.15 mg/kg after 2 weeks once daily administration. The estimated exposure for later- onset SMA patients (2-25 years old at enrolment) in the SUNFISH (Part 2) study at the therapeutic dose (0.25 mg/kg once daily for patients with a body weight <20 kg; 5 mg once daily for patients with a body weight ≥20 kg) was 2070 ng.h/mL after 1 year of treatment and 1940 ng.h/mL after 5 years of treatment. The estimated exposure (mean AUC0-24h) for SMA treatment non-naïve patients (age 1-60 years at enrolment) was 1700 ng.h/mL at the therapeutic dose of 0.25 mg/kg or 5 mg. The observed maximum concentration (mean Cmax) was 194 ng/mL at 0.2 mg/kg in FIREFISH, 140 ng/mL in SUNFISH Part 2, 129 ng/mL in JEWELFISH, and the estimated maximum concentration at 0.15 mg/kg in RAINBOWFISH is 111 ng/mL.
Absorption
Risdiplam was rapidly absorbed in the fasted state with a plasma t max ranging from 1 to 4 hours after oral administration. Based on limited data (n=3), food (high-fat, high calorie breakfast) had no relevant effect on the exposure of risdiplam. In the clinical studies, risdiplam was administered with a morning meal or after breastfeeding.
Distribution
Risdiplam distributes evenly to all parts of the body, including the central nervous system (CNS) by crossing the blood brain barrier, and thereby leading to SMN protein increase in the CNS and throughout the body. Concentrations of risdiplam in plasma and SMN protein in blood reflect its distribution and pharmacodynamic effects in tissues such as brain and muscle.
The population pharmacokinetic parameter estimates were 98 L for the apparent central volume of distribution, 93 L for the peripheral volume, and 0.68 L/hour for the inter-compartment clearance.
Risdiplam is predominantly bound to serum albumin, without any binding to alpha-1 acid glycoprotein, with a free fraction of 11%.
Biotransformation
Risdiplam is primarily metabolized by FMO1 and FMO3, and also by CYPs 1A1, 2J2, 3A4 and 3A7.
Co-administration of 200 mg itraconazole twice daily, a strong CYP3A inhibitor, with a single oral dose of 6 mg risdiplam showed no clinically relevant effect on the PK of risdiplam (11% increase in AUC, 9% decrease in Cmax).
Elimination
Population PK analyses estimated an apparent clearance (CL/F) of 2.6 L/h for risdiplam. The effective half-life of risdiplam was approximately 50 hours in SMA patients.
Risdiplam is not a substrate of human multidrug resistance protein 1 (MDR1).
Approximately 53% of the dose (14% unchanged risdiplam) was excreted in the feces and 28% in urine (8% unchanged risdiplam). Parent drug was the major component found in plasma, accounting for 83% of drug related material in circulation. The pharmacologically inactive metabolite M1 was identified as the major circulating metabolite.
Pharmacokinetics in special populations
Paediatric population
Body weight and age were identified as covariates in the population PK analysis. On the basis of such model, the dose is therefore adjusted based on age (below and above 2 months and 2 years) and body weight (up to 20 kg) to obtain similar exposure across the age and body weight range. Limited PK data are available in patients less than 20 days of age, since only one 16-day-old neonate received risdiplam at a lower dose (0.04 mg/kg) in clinical studies.
Elderly population
No dedicated studies have been conducted to investigate PK in patients with SMA above 60 years of age. Subjects without SMA up to 69 years of age were included in the clinical PK studies, which indicates that no dose adjustment is required for patients up to 69 years of age.
Renal impairment
No studies have been conducted to investigate the PK of risdiplam in patients with renal impairment. Elimination of risdiplam as unchanged entity via renal excretion is minor (8%).
Hepatic impairment
Mild and moderate hepatic impairment had no significant impact on the PK of risdiplam. After a single oral administration of 5 mg risdiplam, the mean ratios for Cmax and AUC were 0.95 and 0.80 in mild (n=8) and 1.20 and 1.08 in moderate hepatic impaired subjects (n=8) versus matched healthy controls (n=10). The safety and PK in patients with severe hepatic impairment have not been studied.
Ethnicity
The PK of risdiplam do not differ in Japanese and Caucasian subjects.
5.3. Preclinical safety data
Impairment of fertility
Treatment with risdiplam was associated with male germ cell arrest in rats and monkeys without safety margins based on systemic exposures at the no observed adverse effect level (NOAEL). These effects led to degenerated spermatocytes, degeneration/necrosis of the seminiferous epithelium, and oligo/aspermia in the epididymis. Sperm cell effects of risdiplam are likely related to an interference of risdiplam with the cell cycle of dividing cells, which is stage specific and expected to be reversible. No effects were seen on female reproductive organs in rats and monkeys after treatment with risdiplam.
No fertility and early embryonic development studies were conducted with concomitant administration of risdiplam, as sperm cell arrest and embryotoxic potential under treatment was already identified with treatment of rats and monkeys in other toxicity studies. No impairment on male fertility or female fertility was observed in two studies in which rats were mated, either following completion of a 13-week treatment period starting at weaning, or 8 weeks after completion of a 4-week treatment period starting at 4 days of age.
Effect on retinal structure
Chronic treatment of monkeys with risdiplam yielded evidence for an effect on the retina in terms of photoreceptor degeneration starting in the periphery of the retina. Upon cessation of treatment, the effects on the retinogram were partially reversible but the photoreceptor degeneration did not reverse. The effects were monitored by optical coherence tomography (OCT) and by electroretinography (ERG). Effects were seen with exposures in excess of 2-fold the exposure in humans at the therapeutic dose without safety margin based on systemic exposures at the NOAEL. No such findings were observed in albino or pigmented rats when dosed chronically with risdiplam at exposures exceeding those in the monkey. Such findings have not been observed in clinical trials in SMA patients with regular ophthalmological monitoring (including SD OCT and visual function assessment).
Effect on epithelial tissues
Effects on skin, larynx and eyelid histology and the gastro intestinal tract were evident in rats and monkeys treated with risdiplam. Changes started to be seen at high doses with treatment of 2 weeks and longer. With chronic treatment for 39 weeks in monkeys, the NOAEL was at an exposure in excess of 2-fold the average exposure in humans at the therapeutic dose.
Effect on haematological parameters
In the acute bone marrow micronucleus test in rats, a reduction of more than 50% in the ratio of polychromatic (young) to normochromatic (adult) erythrocytes, indicative of substantial bone marrow toxicity, was observed at the high dose level with exposure in excess of 15-times the average exposure in humans at the therapeutic dose. With longer treatment of rats for 26 weeks, the exposure margins to the NOAEL were approximately 4-foldthe average exposure in humans at the therapeutic dose.
Genotoxicity
Risdiplam is not mutagenic in a bacterial reverse mutation assay. In mammalian cells in vitro and in bone marrow of rats, risdiplam increases the frequency of micronucleated cells. Micronucleus induction in bone marrow was observed in several toxicity studies in rats (adult and juvenile animals). The NOAEL across the studies is associated with an exposure of approximately 1.5-fold the exposure in humans at the therapeutic dose. Data indicated that this effect is indirect and secondary to an interference of risdiplam with the cell cycle of dividing cells. Risdiplam does not possess a potential to damage DNA directly.
Reproductive toxicity
In studies in pregnant rats treated with risdiplam, embryofoetal toxicity with lower fetal weight and delayed development was evident. The NOAEL for this effect was approximately 2-fold above the exposure levels reached at the therapeutic dose of risdiplam in patients. In studies with pregnant rabbits, dysmorphogenic effects were observed at exposures also associated with maternal toxicity. These consisted of four fetuses (4%) from 4 litters (22%) with hydrocephaly. The NOAEL was approximately 4-fold the exposure levels reached at the therapeutic dose of risdiplam in patients. In a pre- and post-natal development study in rats treated daily with risdiplam, risdiplam caused a slight delay in gestation length. Studies in pregnant and lactating rats showed that risdiplam crosses the placental barrier and is excreted into milk.
Carcinogenicity
Risdiplam did not reveal a carcinogenic potential in transgenic rasH2 mice over 6 months and in a 2 year study in rats at equivalent exposures to those in humans receiving the maximum recommended human dose (MRHD). Significantly increased tumours of the preputial gland in male rats and clitoral gland in female rats at 4 times the exposure of the MRHD are without human relevance, because both are rodent-specific organs.
Juvenile animal studies
Juvenile animal data reveal no special hazard for humans.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.