EZOLETA Tablet Ref.[51159] Active ingredients: Ezetimibe
Source: Medicines Authority (MT) Revision Year: 2022 Publisher: TAD Pharma GmbH, Heinz-Lohmann-Straße 5, 27472 Cuxhaven, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: lipid modifying agents, other lipid modifying agents
ATC code: C10AX09
Mechanism of action
Ezetimibe is in a new class of lipid-lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related plant sterols. Ezetimibe is orally active, and has a mechanism of action that differs from other classes of cholesterol-reducing compounds (e.g. statins, bile acid sequestrants [resins], fibric acid derivatives, and plant stanols). The molecular target of ezetimibe is the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is responsible for the intestinal uptake of cholesterol and phytosterols.
Ezetimibe localises at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver; statins reduce cholesterol synthesis in the liver and together these distinct mechanisms provide complementary cholesterol reduction. In a 2-week clinical study in 18 hypercholesterolemic patients, ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo.
Pharmacodynamic effects
A series of preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of [14C]-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or fat soluble vitamins A and D.
Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C.
Administration of ezetimibe with a statin is effective in reducing the risk of cardiovascular events in patients with coronary heart disease and ACS event history.
Clinical efficacy and safety
In controlled clinical studies, ezetimibe, either as monotherapy or co-administered with a statin significantly reduced total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo B), and triglycerides (TG) and increased high-density lipoprotein cholesterol (HDL-C) in patients with hypercholesterolaemia.
Primary hypercholesterolaemia
In a double-blind, placebo-controlled, 8-week study, 769 patients with hypercholesterolaemia already receiving statin monotherapy and not at National Cholesterol Education Program (NCEP) LDL-C goal (2.6 to 4.1 mmol/l [100 to 160 mg/dl], depending on baseline characteristics) were randomised to receive either ezetimibe 10 mg or placebo in addition to their on-going statin therapy.
Among statin-treated patients not at LDL-C goal at baseline (~82%), significantly more patients randomised to ezetimibe achieved their LDL-C goal at study endpoint compared to patients randomised to placebo, 72% and 19% respectively. The corresponding LDL-C reductions were significantly different (25% and 4% for ezetimibe versus placebo, respectively). In addition, ezetimibe, added to on-going statin therapy, significantly decreased total-C, Apo B, TG and increased HDL-C, compared with placebo. Ezetimibe or placebo added to statin therapy reduced median Creactive protein by 10% or 0% from baseline, respectively.
In two, double-blind, randomised placebo-controlled, 12-week studies in 1.719 patients with primary hypercholesterolaemia, ezetimibe 10 mg significantly lowered total-C (13%), LDL-C (19%), Apo B (14%), and TG (8%) and increased HDL-C (3%) compared to placebo. In addition, ezetimibe had no effect on the plasma concentrations of the fat-soluble vitamins A, D, and E, no effect on prothrombin time, and, like other lipid-lowering agents, did not impair adrenocortical steroid hormone production.
In a multicenter, double-blind, controlled clinical study (ENHANCE), 720 patients with heterozygous familial hypercholesterolemia were randomized to receive ezetimibe 10 mg in combination with simvastatin 80 mg (n = 357) or simvastatin 80 mg (n = 363) for 2 years. The primary objective of the study was to investigate the effect of the ezetimibe/simvastatin combination therapy on carotid artery intima-media thickness (IMT) compared to simvastatin monotherapy. The impact of this surrogate marker on cardiovascular morbidity and mortality is still not demonstrated.
The primary endpoint, the change in the mean IMT of all six carotid segments, did not differ significantly (p= 0.29) between the two treatment groups as measured by B-mode ultrasound. With ezetimibe 10 mg in combination with simvastatin 80 mg or simvastatin 80 mg alone, intima-medial thickening increased by 0.0111 mm and 0.0058 mm, respectively, over the study’s 2 year duration (baseline mean carotid IMT 0.68 mm and 0.69 mm respectively.
Ezetimibe 10 mg in combination with simvastatin 80 mg lowered LDL-C, total-C, Apo B, and TG significantly more than simvastatin 80 mg. The percent increase in HDL-C was similar for the two treatment groups. The adverse reactions reported for ezetimibe 10 mg in combination with simvastatin 80 mg were consistent with its known safety profile.
Paediatric population
In a multicentre, double-blind, controlled study, 138 patients (59 boys and 79 girls), 6 to 10 years of age (mean age 8.3 years) with heterozygous familial or non-familial hypercholesterolaemia (HeFH) with baseline LDL-C levels between 3.74 and 9.92 mmol/l were randomised to either ezetimibe 10 mg or placebo for 12 weeks.
At week 12, ezetimibe significantly reduced total-C (-21% vs. 0%), LDL-C (-28% vs. -1%), Apo-B (-22% vs. -1%), and non-HDL-C (-26% vs. 0%) compared to placebo. Results for the two treatment groups were similar for TG and HDL-C (-6% vs. +8%, and +2% vs. +1%, respectively).
In a multicentre, double-blind, controlled study, 142 boys (Tanner stage II and above) and 106 postmenarchal girls, 10 to 17 years of age (mean age 14.2 years) with heterozygous familial hypercholesterolaemia (HeFH) with baseline LDL-C levels between 4.1 and 10.4 mmol/l were randomised to either ezetimibe 10 mg co-administered with simvastatin (10, 20 or 40 mg) or simvastatin (10, 20 or 40 mg) alone for 6 weeks, co-administered ezetimibe and 40 mg simvastatin or 40 mg simvastatin alone for the next 27 weeks, and open-label co-administered ezetimibe and simvastatin (10 mg, 20 mg, or 40 mg) for 20 weeks thereafter.
At Week 6, ezetimibe co-administered with simvastatin (all doses) significantly reduced total-C (38% vs 26%), LDL-C (49% vs 34%), Apo B (39% vs 27%), and non-HDL-C (47% vs 33%) compared to simvastatin (all doses) alone. Results for the two treatment groups were similar for TG and HDL-C (-17% vs -12% and +7% vs +6%, respectively). At Week 33, results were consistent with those at Week 6 and significantly more patients receiving ezetimibe and 40 mg simvastatin (62%) attained the NCEP AAP ideal goal (<2.8 mmol/L [110 mg/dL]) for LDL-C compared to those receiving 40 mg simvastatin (25%). At Week 53, the end of the open label extension, the effects on lipid parameters were maintained.
The safety and efficacy of ezetimibe co-administered with doses of simvastatin above 40 mg daily have not been studied in paediatric patients 10 to 17 years of age. The safety and efficacy of ezetimibe co-administered with simvastatin have not been studied in paediatric patients <10 years of age.
The long-term efficacy of therapy with ezetimibe in patients below 17 years of age to reduce morbidity and mortality in adulthood has not been studied.
Prevention of Cardiovascular Events
The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) was a multicenter, randomised, double-blind, active-control study of 18,144 patients enrolled within 10 days of hospitalisation for acute coronary syndrome (ACS; either acute myocardial infarction [MI] or unstable angina [UA]). Patients had an LDL-C ≤125 mg/dL (≤3.2 mmol/L) at the time of presentation with ACS if they had not been taking lipid-lowering therapy, or ≤100 mg/dL (≤2.6 mmol/L) if they had been receiving lipid-lowering therapy. All patients were randomised in a 1:1 ratio to receive either ezetimibe/simvastatin 10/40 mg (n=9067) or simvastatin 40 mg (n=9077) and followed for a median of 6.0 years.
Patients had a mean age of 63.6 years; 76% were male, 84% were Caucasian, and 27% were diabetic. The average LDL-C value at the time of study qualifying event was 80 mg/dL (2.1 mmol/L) for those on lipid-lowering therapy (n=6390) and 101 mg/dL (2.6 mmol/L) for those not on previous lipidlowering therapy (n=11594). Prior to the hospitalisation for the qualifying ACS event, 34% of the patients were on statin therapy. At one year, the average LDL-C for patients continuing on therapy was 53.2 mg/dL (1.4 mmol/L) for the ezetimibe/simvastatin group and 69.9 mg/dL (1.8 mmol/L) for the simvastatin monotherapy group. Lipid values were generally obtained for patients who remained on study therapy.
The primary endpoint was a composite consisting of cardiovascular death, major coronary events (MCE; defined as non-fatal myocardial infarction, documented unstable angina that required hospitalisation, or any coronary revascularisation procedure occurring at least 30 days after randomised treatment assignment) and non-fatal stroke. The study demonstrated that treatment with ezetimibe when added to simvastatin provided incremental benefit in reducing the primary composite endpoint of cardiovascular death, MCE, and non-fatal stroke compared with simvastatin alone (relative risk reduction of 6.4%, p=0.016). The primary endpoint occurred in 2572 of 9067 patients (7-year Kaplan-Meier [KM] rate 32.72%) in the ezetimibe/simvastatin group and 2742 of 9077 patients (7-year KM rate 34.67%) in the simvastatin alone group. (See Figure 1 and Table 1.) This incremental benefit is expected to be similar with coadministration of other statins shown to be effective in reducing the risk of cardiovascular events. Total mortality was unchanged in this high risk group (see Table 1).
There was an overall benefit for all strokes; however there was a small non-significant increase in haemorrhagic stroke in the ezetimibe-simvastatin group compared with simvastatin alone (see Table 1). The risk of haemorrhagic stroke for ezetimibe coadministered with higher potency statins in longterm outcome studies has not been evaluated.
The treatment effect of ezetimibe/simvastatin was generally consistent with the overall results across many subgroups, including sex, age, race, medical history of diabetes mellitus, baseline lipid levels, prior statin therapy, prior stroke, and hypertension.
Figure 1. Effect of Ezetimibe/Simvastatin on the Primary Composite Endpoint of Cardiovascular Death, Major Coronary Event, or Non-fatal Stroke:
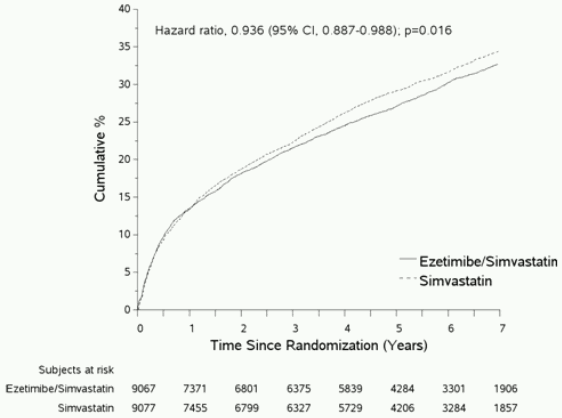
Table 1. Major Cardiovascular Events by Treatment Group in All Randomized Patients in IMPROVE-IT:
| Outcome | Ezetimibe/Simvastatin 10/40 mga (N=9067) | Simvastatin 40 mgb (N=9077) | Hazard Ratio (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| n | K-M %c | n | K-M %c | |||
| Primary Composite Efficacy Endpoint | ||||||
| (CV death, Major Coronary Events and non-fatal stroke) | 2572 | 32.72% | 2742 | 34.67% | 0.936 (0.887, 0.988) | 0.016 |
| Secondary Composite Efficacy Endpoints | ||||||
| CHD death, nonfatal MI, urgent coronary revascularization after 30 days | 1322 | 17.52% | 1448 | 18.88% | 0.912 (0.847, 0.983) | 0.016 |
| MCE, non-fatal stroke, death (all causes) | 3089 | 38.65% | 3246 | 40.25% | 0.948 (0.903, 0.996) | 0.035 |
| CV death, non-fatal MI, unstable angina requiring hospitalization, any revascularization, non-fatal stroke | 2716 | 34.49% | 2869 | 36.20% | 0.945 (0.897, 0.996) | 0.035 |
| Components of Primary Composite Endpoint and Select Efficacy Endpoints (first occurrences of specified event at any time) | ||||||
| Cardiovascular death | 537 | 6.89% | 538 | 6.84% | 1.000 (0.887, 1.127) | 0.997 |
| Major Coronary Event: | ||||||
| Non-fatal MI | 945 | 12.77% | 1083 | 14.41% | 0.871 (0.798, 0.950) | 0.002 |
| Unstable angina requiring hospitalization | 156 | 2.06% | 148 | 1.92% | 1.059 (0.846, 1.326) | 0.618 |
| Coronary revascularization after 30 days | 1690 | 21.84% | 1793 | 23.36% | 0.947 (0.886, 1.012) | 0.107 |
| Non-fatal stroke | 245 | 3.49% | 305 | 4.24% | 0.802 (0.678, 0.949) | 0.010 |
| All MI (fatal and non-fatal) | 977 | 13.13% | 1118 | 14.82% | 0.872 (0.800, 0.950) | 0.002 |
| All stroke (fatal and non-fatal) | 296 | 4.16% | 345 | 4.77% | 0.857 (0.734, 1.001) | 0.052 |
| Non-hemorrhagic stroked | 242 | 3.48% | 305 | 4.23% | 0.793 (0.670, 0.939) | 0.007 |
| Hemorrhagic stroke | 59 | 0.77% | 43 | 0.59% | 1.377 (0.930, 2.040) | 0.110 |
| Death from any cause | 1215 | 15.36% | 1231 | 15.28% | 0.989 (0.914, 1.070) | 0.782 |
a 6% were uptitrated to ezetimibe/simvastatin 10/80 mg.
b 27% were uptitrated to simvastatin 80 mg.
c Kaplan-Meier estimate at 7 years.
d includes ischemic stroke or stroke of undetermined type.
Prevention of Major Vascular Events in Chronic Kidney Disease (CKD)
The Study of Heart and Renal Protection (SHARP) was a multi-national, randomized, placebocontrolled, double-blind study conducted in 9438 patients with chronic kidney disease, a third of whom were on dialysis at baseline. A total of 4650 patients were allocated to a fixed dose combination of ezetimibe 10 mg with simvastatin 20 mg and 4620 to placebo, and followed for a median of 4.9 years. Patients had a mean age of 62 and 63% were male, 72% Caucasian, 23% diabetic and, for those not on dialysis, the mean estimated glomerular filtration rate (eGFR) was 26.5 ml/min/1.73 m². There were no lipid entry criteria. Mean LDL-C at baseline was 108 mg/dL. After one year, including patients no longer taking study medication, LDL-C was reduced 26% relative to placebo by simvastatin 20 mg alone and 38% by ezetimibe 10 mg combined with simvastatin 20 mg.
The SHARP protocol-specified primary comparison was an intention-to-treat analysis of “major vascular events” (MVE; defined as nonfatal MI or cardiac death, stroke, or any revascularization procedure) in only those patients initially randomized to the ezetimibe combined with simvastatin (n=4193) or placebo (n=4191) groups. Secondary analyses included the same composite analyzed for the full cohort randomized (at study baseline or at year 1) to ezetimibe combined with simvastatin (n=4650) or placebo (n=4620) as well as the components of this composite.
The primary endpoint analysis showed that ezetimibe combined with simvastatin significantly reduced the risk of major vascular events (749 patients with events in the placebo group vs. 639 in the ezetimibe combined with simvastatin group) with a relative risk reduction of 16% (p=0.001).
Nevertheless, this study design did not allow for a separate contribution of the monocomponent ezetimibe to efficacy to significantly reduce the risk of major vascular events in patients with CKD.
The individual components of MVE in all randomized patients are presented in Table 2. Ezetimibe combined with simvastatin significantly reduced the risk of stroke and any revascularization, with non-significant numerical differences favouring ezetimibe combined with simvastatin for nonfatal MI and cardiac death.
Table 2. Major Vascular Events by Treatment Group in all randomized patients in SHARPa:
| Outcome | Ezetimibe 10 mg combined with simvastatin 20 mg (N=4650) | Placebo (N=4620) | Risk Ratio (95% CI) | P-value |
|---|---|---|---|---|
| Major Vascular Events | 701 (15.1%) | 814 (17.6%) | 0.85 (0.77-0.94) | 0.001 |
| Nonfatal MI | 134 (2.9%) | 159 (3.4%) | 0.84 (0.66-1.05) | 0.12 |
| Cardiac Death | 253 (5.4%) | 272 (5.9%) | 0.93 (0.78-1.10) | 0.38 |
| Any Stroke | 171 (3.7%) | 210 (4.5%) | 0.81 (0.66-0.99) | 0.038 |
| Non-hemorrhagic Stroke | 131 (2.8%) | 174 (3.8%) | 0.75 (0.60-0.94) | 0.011 |
| Hemorrhagic Stroke | 45 (1.0%) | 37 (0.8%) | 1.21 (0.78-1.86) | 0.40 |
| Any Revascularization | 284 (6.1%) | 352 (7.6%) | 0.79 (0.68-0.93) | 0.004 |
| Major Atherosclerotic Events (MAE)b | 526 (11.3%) | 619 (13.4%) | 0.83 (0.74-0.94) | 0.002 |
a Intention-to-treat analysis on all SHARP patients randomized to ezetimibe combined with simvstatin or placebo either at baseline or year 1
b MAE; defined as the composite of nonfatal myocardial infarction, coronary death, non-hemorrhagic stroke, or any revascularization
The absolute reduction in LDL cholesterol achieved with ezetimibe combined with simvastatin was lower among patients with a lower baseline LDL-C (<2.5 mmol/l) and patients on dialysis at baseline than the other patients, and the corresponding risk reductions in these two groups were attenuated.
Homozygous Familial Hypercholesterolaemia (HoFH)
A double-blind, randomised, 12-week study enrolled 50 patients with a clinical and/or genotypic diagnosis of HoFH, who were receiving atorvastatin or simvastatin (40 mg) with or without concomitant LDL apheresis. Ezetimibe co-administered with atorvastatin (40 or 80 mg) or simvastatin (40 or 80 mg), significantly reduced LDL-C by 15% compared with increasing the dose of simvastatin or atorvastatin monotherapy from 40 to 80 mg.
Homozygous Sitosterolaemia (Phytosterolaemia)
In a double-blind, placebo-controlled, 8-week trial, 37 patients with homozygous sitosterolaemia were randomised to receive ezetimibe 10 mg (n=30) or placebo (n=7). Some patients were receiving other treatments (e.g., statins, resins). Ezetimibe significantly lowered the two major plant sterols, sitosterol and campesterol, by 21% and 24% from baseline, respectively. The effects of decreasing sitosterol on morbidity and mortality in this population are not known.
Aortic Stenosis
The Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis (SEAS) study was a multi-center, double-blind, placebo-controlled study with a median duration of 4.4 years conducted in 1873 patients with asymptomatic aortic stenosis (AS), documented by Doppler-measured aortic peak flow velocity within the range of 2.5 to 4.0 m/s. Only patients who were considered not to require statin treatment for purposes of reducing atherosclerotic cardiovascular disease risk were enrolled. Patients were randomised 1:1 to receive placebo or co-administered ezetimibe 10 mg and simvastatin 40 mg daily.
The primary endpoint was the composite of major cardiovascular events (MCE) consisting of cardiovascular death, aortic valve replacement (AVR) surgery, congestive heart failure (CHF) as a result of progression of AS, nonfatal myocardial infarction, coronary artery bypass grafting (CABG), percutaneous coronary intervention (PCI), hospitalization for unstable angina, and nonhemorrhagic stroke. The key secondary endpoints were composites of subsets of the primary endpoint event categories.
Compared to placebo, ezetimibe/simvastatin 10/40 mg did not significantly reduce the risk of MCE.
The primary outcome occurred in 333 patients (35.3%) in the ezetimibe/simvastatin group and in 355 patients (38.2%) in the placebo group (hazard ratio in the ezetimibe/simvastatin group, 0.96; 95% confidence interval, 0.83 to 1.12; p=0.59). Aortic valve replacement was performed in 267 patients (28.3%) in the ezetimibe/simvastatin group and in 278 patients (29.9%) in the placebo group (hazard ratio, 1.00; 95% CI, 0.84 to 1.18; p=0.97). Fewer patients had ischemic cardiovascular events in the ezetimibe/simvastatin group (n=148) than in the placebo group (n=187) (hazard ratio, 0.78; 95% CI, 0.63 to 0.97; p=0.02), mainly because of the smaller number of patients who underwent coronary artery bypass grafting.
Cancer occurred more frequently in the ezetimibe/simvastatin group (105 versus 70, p=0.01). The clinical relevance of this observation is uncertain as in the bigger SHARP trial the total number of patients with any incident cancer (438 in the ezetimibe/simvastatin versus 439 placebo group) did not differ. In addition, in the IMPROVE-IT trial the total number of patients with any new malignancy (853 in the ezetimibe/simvastatin group versus 863 in the simvastatin group) did not differ significantly and therefore the finding of SEAS trial could not be confirmed by SHARP or IMPROVE-IT.
5.2. Pharmacokinetic properties
Absorption
After oral administration, ezetimibe is rapidly absorbed and extensively conjugated to a pharmacologically-active phenolic glucuronide (ezetimibe-glucuronide). Mean maximum plasma concentrations (Cmax) occur within 1 to 2 hours for ezetimibe-glucuronide and 4 to 12 hours for ezetimibe. The absolute bioavailability of ezetimibe cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection.
Concomitant food administration (high fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe when administered as ezetimibe 10-mg tablets. Ezoleta can be administered with or without food.
Distribution
Ezetimibe and ezetimibe glucuronide are bound 99.7% and 88 to 92% to human plasma proteins, respectively.
Biotransformation
Ezetimibe is metabolised primarily in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are slowly eliminated from plasma with evidence of significant enterohepatic recycling. The half-life for ezetimibe and ezetimibe-glucuronide is approximately 22 hours.
Elimination
Following oral administration of 14C ezetimibe (20 mg) to human subjects, total ezetimibe accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the faeces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Special populations
Paediatric population
The pharmacokinetics of ezetimibe are similar between children ≥6 years and adults. Pharmacokinetic data in the paediatric population <6 years of age are not available. Clinical experience in paediatric and adolescent patients includes patients with HoFH, HeFH, or sitosterolaemia.
Elderly
Plasma concentrations for total ezetimibe are about 2-fold higher in the elderly (more than 65 years) than in the young (18 to 45 years). LDL-C reduction and safety profile are comparable between elderly and young subjects treated with Ezoleta. Therefore, no dosage adjustment is necessary in the elderly.
Hepatic impairment
After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh score 5 or 6), compared to healthy subjects. In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment (Child-Pugh score 7 to 9), the mean AUC for total ezetimibe was increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects. No dosage adjustment is necessary for patients with mild hepatic impairment. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe (Child-Pugh score >9) hepatic impairment, Ezoleta is not recommended in these patients (see section 4.4).
Renal impairment
After a single 10 mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl less than 0.5 ml/s/1.73 m² (30 ml/min/1.73 m²)), the mean AUC for total ezetimibe was increased approximately 1.5-fold, compared to healthy subjects (n=9). This result is not considered clinically significant. No dosage adjustment is necessary for renally impaired patients.
An additional patient in this study (post-renal transplant and receiving multiple medications, including ciclosporin) had a 12-fold greater exposure to total ezetimibe.
Gender
Plasma concentrations for total ezetimibe are slightly higher (approximately 20%) in women than in men. LDL-C reduction and safety profile are comparable between men and women treated with ezetimibe. Therefore, no dosage adjustment is necessary on the basis of gender.
5.3. Preclinical safety data
Animal studies on the chronic toxicity of ezetimibe identified no target organs for toxic effects. In dogs treated for four weeks with ezetimibe (0.03 mg/kg/day) the cholesterol concentration in the cystic bile was increased by a factor of 2.5 to 3.5. However, in a one-year study on dogs given doses of up to 300 mg/kg/day no increased incidence of cholelithiasis or other hepatobiliary effects were observed. The significance of these data for humans is not known. A lithogenic risk associated with the therapeutic use of ezetimibe cannot be ruled out.
In co-administration studies with ezetimibe and statins the toxic effects observed were essentially those typically associated with statins. Some of the toxic effects were more pronounced than observed during treatment with statins alone. This is attributed to pharmacokinetic and pharmacodynamic interactions in co-administration therapy. No such interactions occurred in the clinical studies. Myopathy occurred in rats only after exposure to doses that were several times higher than the human therapeutic dose (approximately 20 times the AUC level for statins and 500 to 2.000 times the AUC level for the active metabolites).
In a series of in vivo and in vitro assays ezetimibe, given alone or co-administered with statins, exhibited no genotoxic potential. Long-term carcinogenicity tests on ezetimibe were negative. Ezetimibe had no effect on the fertility of male or female rats, nor was it teratogenic in rats or rabbits, nor did it affect prenatal or postnatal development. Ezetimibe crossed the placental barrier in pregnant rats and rabbits given multiple doses of 1.000 mg/kg/day. The co-administration of ezetimibe and statins was not teratogenic in rats. In pregnant rabbits a small number of skeletal deformities (fused thoracic and caudal vertebrae, reduced number of caudal vertebrae) were observed. The co-administration of ezetimibe with lovastatin resulted in embryolethal effects.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.