FILSPARI Film-coated tablet Ref.[110236] Active ingredients: Sparsentan
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Vifor France, 100–101 Terrasse Boieldieu, Tour Franklin La Défense 8, 92042 Paris La Défense Cedex, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: agents acting on the renin-angiotensin system
ATC code: C09XX01
Mechanism of action
Sparsentan is a dual endothelin angiotensin receptor antagonist.
It is a single molecule that functions as a high affinity, dual-acting antagonist of both the ETAR and AT1R. Endothelin 1, via ETAR, and angiotensin II, via AT1R, mediate processes that lead to IgAN progression through haemodynamic actions and mesangial cell proliferation, increased expression and activity of proinflammatory and profibrotic mediators, podocyte injury, and oxidative stress. Sparsentan inhibits activation of both ETAR and AT1R and thereby reduces proteinuria and slows the progression of kidney disease.
Pharmacodynamic effects
In a randomised, positive- and placebo-controlled study with healthy subjects, sparsentan caused mild QTcF prolongation with a peak effect of 8.8 ms (90% CI: 5.9, 11.8) at 800 mg and 8.1 ms (5.2, 11.0) at 1600 mg. In an additional study with healthy subjects, at sparsentan exposure exceeding exposure at maximum recommended human dose by more than 2-fold, the peak effect was 8.3 (6.69, 9.90) ms. Therefore, it is unlikely that sparsentan has a clinically relevant effect on QT prolongation.
Clinical efficacy and safety
The efficacy and safety of sparsentan has been evaluated in PROTECT in patients with IgAN.
PROTECT is a randomised, double-blind (110 weeks), active-controlled, multicentre, global phase 3 trial in patients with IgAN. The trial enrolled patients aged ≥18 years, including 15 (8%) sparsentan-treated patients aged >65 years, with an eGFR ≥30 mL/min/1.73 m² and total urine protein excretion ≥1.0 g/day. Prior to enrolment, patients were on the maximum tolerated dose of an ACE inhibitor and/or an ARB for at least 3 months. The ACE inhibitors and/or ARB therapy were discontinued prior to initiation of sparsentan. Patients with a baseline potassium value exceeding 5.5 mmol/L were excluded.
A total of 404 patients were randomised and received sparsentan (n=202) or irbesartan (n=202). Treatment was initiated with sparsentan at 200 mg once daily or irbesartan 150 mg once daily. After 14 days, the dose was to be titrated, as tolerated, to the recommended dose of sparsentan 400 mg once daily or irbesartan 300 mg once daily. Dose tolerance was defined as systolic blood pressure >100 mmHg and diastolic blood pressure >60 mmHg after 2 weeks and no AEs (e.g, worsening oedema) or laboratory findings (e.g, serum potassium >5.5 mEq/L [5.5 mmol/L]). Inhibitors of the RAAS or endothelin system were prohibited during the trial. Other classes of antihypertensive agents were permitted as needed to achieve target blood pressure. Treatment with immunosuppressive agents was permitted during the trial at the discretion of the investigator.
Baseline characteristics for eGFR and proteinuria were comparable between treatment groups. The overall population had a mean (SD) eGFR of 57 (24) mL/min/1.73 m² and a median urine protein/creatinine (UP/C) ratio of 1.24 g/g (interquartile range: 0.83, 1.77). The mean age was 46 years (range 18 to 76 years); 70% were male, 67% White, 28% Asian, 1% Black or African American, and 3% were other race.
The primary (interim) analysis of proteinuria was conducted after 36 weeks following randomization of approximately 280 subjects, to determine whether the treatment effect of the primary efficacy endpoint, the change from baseline in UP/C at week 36, is statistically significant. The trial met its primary endpoint, which was change from baseline in the UP/C ratio at week 36. Geometric mean UP/C at week 36 was 0.62 g/g in the sparsentan arm versus 1.07 g/g in the irbesartan arm. The geometric least squares mean percent change in UP/C from baseline at week 36 was -49.8% (95% confidence interval [CI]: -54.98, -43.95) in the sparsentan arm versus -15.1% (95% CI: -23.72, -5.39) in the irbesartan arm (p<0.0001). At the final analysis, sparsentan demonstrated a rapid and durable antiproteinuric treatment effect over 2 years, with a geometric mean UP/C at week 110 of 0.64 g/g in the sparsentan arm versus 1.09 g/g in the irbesartan arm representing a 43% mean reduction from baseline (95% CI: -49.75, -34.97) compared to only 4.4% for irbesartan (95% CI: -15.84, 8.70). Improvement in proteinuria reduction was consistently observed with sparsentan as early as 4 weeks and sustained through week 110 (Figure 1).
Figure 1. Percent change from baseline urine protein/creatinine ratio by visit (PROTECT):
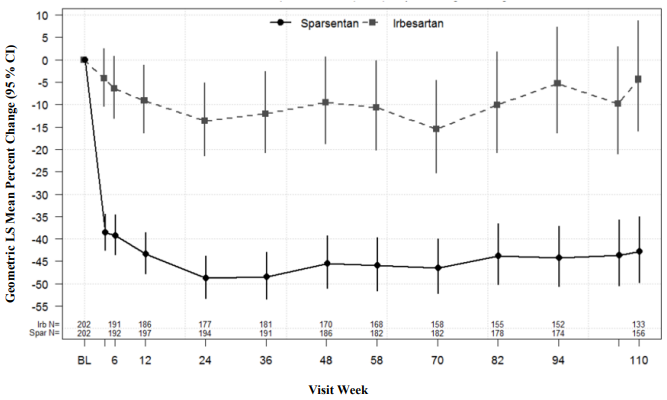
Notes: Adjusted geometric least squares mean ratio of UP/C relative to baseline was based on a longitudinal repeated measures model stratified by screening eGFR and proteinuria, reported as percentage change along with the respective 95% CI. Analysis includes UP/C data during the double-blind period from all patients who were randomised and received at least 1 dose of study medication. Baseline was defined as the last non-missing observation prior to and including the start of dosing.
Abbreviations: CI = confidence interval; eGFR = estimated glomerular filtration rate; LS = least squares; UP/C = urine protein/creatinine ratio
Estimated GFR
At the time of confirmatory analysis, the improvement in 2 year eGFR chronic slope (from 6 weeks onwards) was 1.1 mL/min/1.73 m² per year with sparsentan compared to irbesartan (95% CI: 0.07, 2.12; p=0.037), and the corresponding improvement in 2 year eGFR total slope (from baseline onwards) was 1.0 mL/min/1.73 m² per year (95% CI: -0.03, 1.94; p=0.058). The absolute change from baseline in eGFR at 2 years was -5.8 mL/min/1.73 m² (95% CI: -7.38, -4.24) for sparsentan compared to -9.5 mL/min/1.73 m² (95% CI: -11.17, -7.89) for irbesartan.
Additional information
Two large randomised, controlled trials (ONTARGET (ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) and VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes)) have examined the use of the combination of an ACE-inhibitor with an angiotensin II receptor blocker. ONTARGET was a study conducted in patients with a history of cardiovascular or cerebrovascular disease, or type 2 diabetes mellitus accompanied by evidence of end-organ damage. VA NEPHRON-D was a study in patients with type 2 diabetes mellitus and diabetic nephropathy. These studies have shown no significant beneficial effect on renal and/or cardiovascular outcomes and mortality, while an increased risk of hyperkalaemia, acute kidney injury and/or hypotension as compared to monotherapy was observed. Given their similar pharmacodynamic properties, these results are also relevant for other ACE inhibitors and angiotensin II receptor blockers. ACE inhibitors and angiotensin II receptor blockers should therefore not be used concomitantly in patients with diabetic nephropathy. ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) was a study designed to test the benefit of adding aliskiren to a standard therapy of an ACE inhibitor or an angiotensin II receptor blocker in patients with type 2 diabetes mellitus and chronic kidney disease, cardiovascular disease, or both. The study was terminated early because of an increased risk of adverse outcomes. Cardiovascular death and stroke were both numerically more frequent in the aliskiren group than in the placebo group and adverse events and serious adverse events of interest (hyperkalaemia, hypotension and renal dysfunction) were more frequently reported in the aliskiren group than in the placebo group.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Filspari in one or more subsets of the paediatric population in the treatment of immunoglobulin A nephropathy (see section 4.2 for information on paediatric use).
Conditional approval
This medicinal product has been authorised under a so-called ‘conditional approval’ scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Absorption
Following a single oral dose of 400 mg sparsentan, the median time to peak plasma concentration is approximately 3 hours.
Following a single oral dose of 400 mg sparsentan, the geometric mean Cmax and AUC are 6.97 μg/mL and 83 μg × h/mL, respectively. Steady-state plasma levels are reached within 7 days with no accumulation of exposure at the recommended dosage.
Following a dose of 400 mg sparsentan daily, the steady-state geometric mean Cmax and AUC are 6.47 μg/mL and 63.6 μg × h/mL, respectively.
Food effect
At doses of 400 mg and below, the effect of a high fat meal on sparsentan exposure was not clinically relevant. Sparsentan can be taken with or without food.
Distribution
Based on population pharmacokinetic analysis, the apparent volume of distribution at steady state is 61.4 L.
Sparsentan is highly bound (>99%) to human plasma proteins with preferential binding to albumin and moderate binding to α1-acid glycoprotein.
Biotransformation
Sparsentan is primarily metabolised by CYP3A4 with a minor contribution of CYP2C8, 2C9 and 3A5. Parent compound is the predominant entity in human plasma, representing approximately 90% of the total radioactivity in circulation. A minor hydroxylated metabolite was the only metabolite in plasma that accounted for >1% of the total radioactivity (approximately 3%). The main metabolic pathway of sparsentan was oxidation and dealkylation, and 9 metabolites were identified in human faeces, plasma and urine.
Elimination
The clearance of sparsentan is time dependent. Based on population pharmacokinetic analysis, the apparent clearance is 3.88 L/h, increasing to 5.11 L/h at steady state.
The half-life of sparsentan at steady state is estimated to be 9.6 hours.
Following a single 400 mg dose of radiolabelled sparsentan, 82 % of the dosed radioactivity was recovered within a 10 day collection period: 80 % via the faeces with 9 % as unchanged, and 2 % via the urine with a negligible amount as unchanged.
Linearity/non-linearity
The Cmax and AUC of sparsentan increase less than proportionally following administration of single doses of 200 mg to 1600 mg. Sparsentan showed time-dependent pharmacokinetics with no Cmax accumulation and decreased AUC at steady state following a dose of 400 or 800 mg daily.
Special populations
Elderly
Population pharmacokinetic analysis found no significant effect of age on the plasma exposure of sparsentan. No dosage adjustment is necessary for elderly patients (see section 4.2). Sparsentan has not been studied in patients >75 years of age.
Hepatic impairment
In a dedicated hepatic impairment study, systemic exposure following a single dose of 400 mg sparsentan was similar in patients with baseline mild or moderate hepatic impairment (Child-Pugh A or Child-Pugh B classification) compared to patients with normal hepatic function. No dose adjustment is required in patients with mild or moderate hepatic impairment. Sparsentan should be used with caution in patients with moderate hepatic impairment (see sections 4.2 and 4.4).
No data are available in patients with severe hepatic impairment and sparsentan is therefore not recommended in these patients (Child-Pugh C classification) (see section 4.2).
Renal impairment
Based on population pharmacokinetic analysis in chronic kidney disease patients with mild (creatinine clearance 60 to 89 mL/min), moderate (creatinine clearance 30 to 59 mL/min), and severe (creatinine clearance 15 to 29 mL/min) kidney disease, there is no clinically meaningful effect of kidney impairment on pharmacokinetics as compared to normal kidney function (creatinine clearance ≥90 mL/min). No data are available in patients with end-stage kidney disease (creatinine clearance <15 mL/min).
Based on limited available data, no dose adjustment can be recommended for patients with severe kidney disease (eGFR <30 mL/min/1.73 m², see section 4.2). Sparsentan has not been studied in patients with severe kidney disease or undergoing dialysis, therefore sparsentan is not recommended in these patients. Sparsentan has not been studied in patients who have received a kidney transplant, therefore in this patient population sparsentan should be used with caution (see section 4.2).
Other special populations
Population pharmacokinetic analyses indicate that there is no clinically meaningful effect of age, gender, or race on the pharmacokinetics of sparsentan.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction, and juvenile development.
Adverse reactions not observed in clinical studies but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:
In embryo-foetal development studies in rat and rabbit, developmental toxicity was seen in both species. In rats, dose-dependent teratogenic effects in the form of craniofacial malformations, skeletal abnormalities, increased embryo-foetal lethality, and reduced foetal weights were observed at all doses of sparsentan tested at exposures 8-fold and 13-fold over the AUC for 800 mg/day and 400 mg/day in humans. In rabbits, there were no foetal malformations or effects on embryo-foetal viability or foetal growth, but an increase in skeletal variations (supernumerary cervical ribs) occurred at an exposure of approximately 0.10 and 0.2 times the AUC in humans at 800 mg/day and 400 mg/day.
In the pre- and postnatal development study in rat, maternal toxicity including death was seen at ~8-fold and 13-fold, and maternal toxicity at ~2-fold and 3-fold the AUC in humans at 800 mg/day and 400 mg/day. An increase in pup deaths and decreased growth occurred at ~8-fold and 13-fold, and decreased growth at ~2-fold and 3-fold the AUC in humans at 800 mg/day and 400 mg/day.
Juvenile animal studies
Juvenile animal studies in rats demonstrated that there were no general toxicological adverse effects seen up to 10 mg/kg/day and no reproductive toxicity in males or females up to 60 mg/kg/day when dosing started on postnatal day (PND) 14 (equivalent to 1 year old children). Vascular toxicity occurred at doses ≥3 mg/kg/day when dosing started on PND 7 (equivalent to newborn infants).
Environmental risk assessment (ERA)
Conclusions of studies for sparsentan show that sparsentan is considered not to be persistent, bioaccumulative and toxic (PBT) nor very persistent and very bioaccumulative (vPvB). A risk to the sewage treatment plant, surface water, groundwater, sediment and terrestrial compartment is not anticipated based on the prescribed use of sparsentan (see section 6.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.