FINLEE Dispersible tablet Ref.[51662] Active ingredients: Dabrafenib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors, B-Raf serine-threonine kinase (BRAF) inhibitors
ATC code: L01EC02
Mechanism of action
Dabrafenib is an inhibitor of RAF kinases. Oncogenic mutations in BRAF lead to constitutive activation of the RAS/RAF/MEK/ERK pathway. The most commonly observed BRAF mutation is V600E, which has been identified in 19% of paediatric LGG and approximately 5% of paediatric HGG.
Combination with trametinib
Trametinib is a reversible, highly selective, allosteric inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activation and kinase activity. MEK proteins are components of the extracellular signal-related kinase (ERK) pathway. In human cancers, this pathway is often activated by mutated forms of BRAF which activates MEK. Trametinib inhibits activation of MEK by BRAF and inhibits MEK kinase activity.
Thus, trametinib and dabrafenib inhibit two kinases in this pathway, MEK and RAF, and therefore the combination provides concomitant inhibition of the pathway. The combination of dabrafenib with trametinib has shown anti-tumour activity in BRAF V600 mutation-positive cancer cell lines in vitro and delays the emergence of resistance in vivo in BRAF V600 mutation-positive xenografts.
Pharmacodynamic effects
Preclinical data generated in biochemical assays demonstrated that dabrafenib inhibits BRAF kinases with activating codon 600 mutations (Table 5).
Table 5. Kinase inhibitory activity of dabrafenib against RAF kinases:
| Kinase | Inhibitory concentration 50 (nM) |
|---|---|
| BRAF V600E | 0.65 |
| BRAF WT | 3.2 |
| CRAF WT | 5.0 |
Clinical efficacy and safety
Paediatric population
The clinical efficacy and safety of dabrafenib plus trametinib combination therapy in paediatric patients aged 1 to <18 years with BRAF V600 mutation-positive glioma was evaluated in a multicentre, open-label, Phase II clinical study (EudraCT 2015-004015-20). Patients with low-grade glioma (WHO 2016 Grades 1 and 2) who required first systemic therapy were randomised in a 2:1 ratio to dabrafenib plus trametinib or carboplatin plus vincristine, and patients with relapsed or refractory high-grade glioma (WHO 2016 Grades 3 and 4) were enrolled into a single-arm dabrafenib plus trametinib cohort.
BRAF mutation status was identified prospectively via a local test, or a central laboratory real-time polymerase chain reaction (PCR) test when a local test was not available. In addition, retrospective testing of available tumour samples by the central laboratory was performed to confirm the BRAF V600E mutation.
Dabrafenib and trametinib dosing in the clinical study was age- and weight-dependent, with dabrafenib dosed orally at 2.625 mg/kg twice daily for ages <12 years and at 2.25 mg/kg twice daily for ages 12 years and older; trametinib was dosed orally at 0.032 mg/kg once daily for ages <6 years and at 0.025 mg/kg once daily for ages 6 years and older. Dabrafenib doses were capped at 150 mg twice daily and trametinib doses at 2 mg once daily. Carboplatin and vincristine were dosed based on age and body surface area at doses of 175 mg/m² and 1.5 mg/m², respectively, as weekly infusions. Carboplatin and vincristine were administered in one 10-week induction course followed by eight 6-week cycles of maintenance therapy.
The primary efficacy endpoint in both cohorts was overall response rate (ORR, sum of confirmed complete/CR and partial responses/PR) by independent review based on RANO (2017) criteria for the LGG cohort, and RANO (2010) criteria for the HGG cohort. The primary analysis was performed when all patients in both cohorts had completed at least 32 weeks of therapy.
BRAF mutation-positive paediatric low-grade glioma (WHO Grades 1 and 2)
In the low-grade glioma cohort, 110 patients were randomised to dabrafenib plus trametinib (n=73) or carboplatin plus vincristine (n=37). Median age was 9.5 years, with 34 patients (30.9%) aged 12 months to <6 years, 36 patients (32.7%) aged 6 to <12 years and 40 patients (36.4%) aged 12 to <18 years; 60% were female. The majority of patients (80%) had Grade 1 glioma at initial diagnosis. The most common pathologies were pilocytic astrocytoma (30.9%), ganglioglioma (27.3%) and LGG not otherwise specified (NOS) (18.2%). Metastatic sites were present in 9 patients (8.2%). Prior surgery was reported in 91 patients (82.7%), among those patients the procedure at last surgery was resection in 28 patients (25.5%). Systemic corticosteroid use was reported in 36 patients (32.7%).
The ORR in the dabrafenib plus trametinib arm showed a statistically significant improvement over carboplatin plus vincristine. The subsequent hierarchical testing also demonstrated a statistically significant improvement in progression-free survival (PFS) over chemotherapy (Table 6).
At the time of the primary analysis, conducted after all patients had completed at least 32 weeks of treatment or had discontinued earlier, the overall survival (OS) data were still immature (one death was reported in the carboplatin plus vincristine (C+V) arm).
Table 6. Response and progression-free survival in the pivotal study G2201 (LGG cohort, primary analysis):
| Dabrafenib + Trametinib (D+T) N=73 | Carboplatin + Vincristine (C+V) N=37 | |
|---|---|---|
| Best overall response | ||
| Complete response (CR), n (%) | 2 (2.7) | 1 (2.7) |
| Partial response (PR), n (%) | 32 (43.8) | 3 (8.1) |
| Stable disease (SD), n (%) | 30 (41.1) | 15 (40.5) |
| Progressive disease (PD), n (%) | 8 (11.0) | 12 (32.4) |
| Unknown, n (%) | 1 (1.4) | 6 (16.2)1 |
| Overall response rate | ||
| ORR (CR+PR), 95% CI | 46.6% (34.8 – 58.6%) | 10.8% (3.0 – 25.4%) |
| Odds ratio2 | 7.19 (2.3 – 22.4), p<0.001 | |
| Risk difference | 35.8% (20.6 – 51.0) | |
| Progression-free survival (PFS) | ||
| PFS (months), (95% CI) | 20.1 (12.8 – NE) | 7.4 (3.6 – 11.8) |
| Hazard ratio (95% CI), p-value | 0.31 (0.17 – 0.55), p<0.001 | |
NE=not evaluable
1 4 patients randomised to C+V discontinued prior to receiving treatment.
2 Odds ratio (D+T vs C+V) and 95% CI are from a logistic regression with treatment as the only covariate, i.e. it is the odds of observing a response in the D+T arm compared to the odds of observing a response in the C+V arm. Odds ratio >1 favours D+T.
Figure 1. Kaplan-Meier curves for progression-free survival in the pivotal study G2201 (LGG cohort, primary analysis):
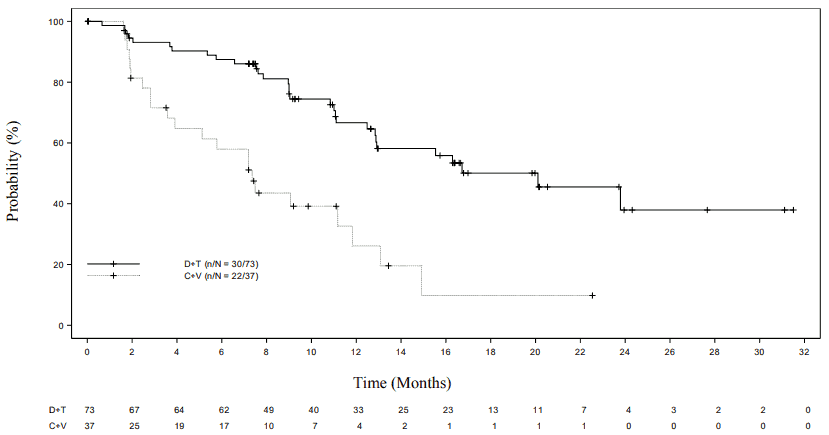
BRAF mutation-positive paediatric high-grade glioma (WHO Grades 3 and 4)
In the single-arm high-grade glioma cohort, 41 patients with relapsed or refractory HGG were enrolled and treated with dabrafenib plus trametinib for a median duration of 72.7 weeks. Median age was 13.0 years, with 5 patients (12.2%) aged 12 months to <6 years, 10 patients (24.4%) aged 6 to <12 years and 26 patients (63.4%) aged 12 to <18 years; 56% were female. The histological grade at initial diagnosis was Grade 4 in 20 patients (48.8%), Grade 3 in 13 patients (31.7%), Grade 2 in 4 patients (9.8%), Grade 1 in 3 patients (7.3%) and missing in 1 patient (2.4%). The most common pathologies were glioblastoma multiforme (31.7%), anaplastic pleomorphic xanthoastrocytoma (14.6%), HGG NOS (9.8%) and pleomorphic xanthoastrocytoma (9.8%). Prior surgery was reported in 40 patients (97.6%), among those patients the procedure at last surgery was resection in 24 patients (58.5%). Prior antineoplastic chemotherapy was reported for 33 patients (80.5%). Prior radiotherapy was reported for 37 patients (90.2%). Systemic corticosteroid use while on study treatment was reported in 21 patients (51.2%).
The ORR in this cohort was 56.1% (23/41), 95% CI (39.7%, 71.5%): CR in 12 patients (29.3%) and PR in 11 patients (26.8%). The median duration of response (DOR) was 22.2 months (95% CI: 7.6 – NE), with 15 patients (65.2%) censored at the time of the primary analysis.
5.2. Pharmacokinetic properties
The pharmacokinetic properties of dabrafenib have mostly been determined in adult patients using the solid (capsule) formulation. The pharmacokinetics of dabrafenib following single or repeat weightadjusted dosing were also evaluated in 243 paediatric patients. The population pharmacokinetic analysis included 61 patients aged 1 to <6 years, 77 patients aged 6 to <12 years and 105 patients aged 12 to <18 years. Clearance was comparable with clearance in adult patients. Weight was identified as a significant covariate of dabrafenib clearance. Age was not a significant additional covariate. The pharmacokinetic exposures of dabrafenib at the recommended weight-adjusted dose in paediatric patients were within range of those observed in adults.
Absorption
The dabrafenib dispersible tablet suspension was absorbed rapidly with a median time to achieve peak plasma concentration of 1.5 hours post-dose. The mean absolute oral bioavailability of dabrafenib capsules was 94.5%. The suspension is expected to have 20% lower bioavailability. Based on data from adult patients with the capsule formulation, a decrease in exposure was observed with repeat dosing, likely due to induction of its own metabolism. Mean accumulation AUC Day 18/Day 1 ratio was 0.73.
Dabrafenib exposure (Cmax and AUC) increased in a dose-proportional manner between 12 mg and 300 mg following single-dose administration, but the increase was less than dose-proportional after repeat twice-daily dosing.
In the pivotal paediatric study, steady-state geometric mean (CV) Cmax and AUCtau were 1330 ng/ml (93.5) and 4910 ng*hr/ml (54.0%) in the LGG cohort and 1520 ng/ml (65.9%) and 4300 ng*hr/ml (44.7%) in the HGG cohort.
Food effect
The impact of food on the pharmacokinetics of the dispersible tablets suspension has not been investigated. Administration of dabrafenib (capsule formulation) with food reduced the bioavailability (Cmax and AUC decreased by 51% and 31% respectively) and delayed the absorption of dabrafenib when compared to the fasted state in an adult healthy volunteer study.
Distribution
Dabrafenib binds to human plasma proteins and is 99.7% bound. The steady-state volume of distribution following intravenous microdose administration in adults was 46 L.
Biotransformation
The metabolism of dabrafenib is primarily mediated by CYP2C8 and CYP3A4 to form hydroxydabrafenib, which is further oxidised via CYP3A4 to form carboxy-dabrafenib. Carboxy-dabrafenib can be decarboxylated via a non-enzymatic process to form desmethyl-dabrafenib. Carboxydabrafenib is excreted in bile and urine. Desmethyl-dabrafenib may also be formed in the gut and reabsorbed. Desmethyl-dabrafenib is metabolised by CYP3A4 to oxidative metabolites. Hydroxydabrafenib terminal half-life parallels that of parent with a half-life of 10 hrs while the carboxy- and desmethyl-metabolites exhibited longer half-lives (21 to 22 hours). In paediatric patients, the mean metabolite-to-parent AUC ratios (% CV) following repeat-dose administration of the capsules or of the dispersible tablet suspension were 0.64 (28%), 15.6 (49%) and 0.69 (62%) for hydroxy-, carboxy-, and desmethyl-dabrafenib, respectively. Based on exposure, relative potency, and pharmacokinetic properties, both hydroxy- and desmethyl-dabrafenib are likely to contribute to the clinical activity of dabrafenib while the activity of carboxy-dabrafenib is not likely to be significant.
Elimination
Terminal half-life of dabrafenib following an intravenous single microdose in adult patients was 2.6 hours. Dabrafenib terminal half-life after a single oral dose of the dispersible tablet formulation was 11.5 hours (CV of 67.7%) in an adult healthy volunteer study. The apparent clearance of dabrafenib in paediatric patients (median body weight: 38.7 kg) was 11.8 L/h (CV of 49%).
After an oral dose, the major route of elimination of dabrafenib is metabolism, mediated via CYP3A4 and CYP2C8. Dabrafenib-related material was excreted primarily in faeces, with 71% of an oral dose recovered in faeces; 23% of the dose was recovered in urine in the form of metabolites only.
Medicinal product interactions
Effects of other medicinal products on dabrafenib
Dabrafenib is a substrate of human P-glycoprotein (P-gp) and human BCRP in vitro. However, these transporters have minimal impact on dabrafenib oral bioavailability and elimination and the risk for clinically relevant drug-drug interactions with inhibitors of P-gp or BCRP is low. Neither dabrafenib nor its 3 main metabolites were demonstrated to be inhibitors of P-gp in vitro.
Effects of dabrafenib on other medicinal products
Although dabrafenib and its metabolites, hydroxy-dabrafenib, carboxy-dabrafenib and desmethyldabrafenib, were inhibitors of human organic anion transporter 1 (OAT1) and OAT3 in vitro, and dabrafenib and its desmethyl-metabolite were found to be inhibitors of organic cation transporter 2 (OCT2) in vitro, the risk of a drug-drug interaction with these transporters is minimal based on clinical exposure of dabrafenib and its metabolites.
Special patient populations
Hepatic impairment
A population pharmacokinetic analysis in adult patients indicates that mildly elevated bilirubin and/or AST levels (based on National Cancer Institute [NCI] classification) do not significantly affect dabrafenib oral clearance. In addition, mild hepatic impairment as defined by bilirubin and AST did not have a significant effect on dabrafenib metabolite plasma concentrations. No data are available in patients with moderate to severe hepatic impairment. As hepatic metabolism and biliary secretion are the primary routes of elimination of dabrafenib and its metabolites, administration of dabrafenib should be undertaken with caution in patients with moderate to severe hepatic impairment (see section 4.2).
Renal impairment
A population pharmacokinetic analysis in adult patients suggests that mild renal impairment does not affect oral clearance of dabrafenib. Although data in moderate renal impairment are limited these data may indicate no clinically relevant effect. No data are available in patients with severe renal impairment (see section 4.2).
Race
A population pharmacokinetic analysis in adult patients showed no significant differences in the pharmacokinetics of dabrafenib between Asian and Caucasian patients. There are insufficient data to evaluate the potential effect of other races on dabrafenib pharmacokinetics.
Gender
Based on population pharmacokinetic analyses in adult and paediatric patients, estimated clearance of dabrafenib was slightly lower in female patients, but the difference was not considered clinically relevant.
5.3. Preclinical safety data
Carcinogenicity studies with dabrafenib have not been conducted. Dabrafenib was not mutagenic or clastogenic using in vitro tests in bacteria and cultured mammalian cells, and an in vivo rodent micronucleus assay.
In combined female fertility, early embryonic and embryo-foetal development studies in rats numbers of ovarian corpora lutea were reduced in pregnant females at 300 mg/kg/day (approximately 3 times human clinical exposure based on AUC), but there were no effects on oestrous cycle, mating or fertility indices. Developmental toxicity including embryo-lethality and ventricular septal defects and variation in thymic shape were seen at 300 mg/kg/day, and delayed skeletal development and reduced foetal body weight at ≥20 mg/kg/day (≥0.5 times human clinical exposure based on AUC).
Male fertility studies with dabrafenib have not been conducted. However, in repeat dose studies, testicular degeneration/depletion was seen in rats and dogs (≥0.2 times human clinical exposure based on AUC). Testicular changes in rat and dog were still present following a 4-week recovery period (see section 4.6).
Cardiovascular effects, including coronary arterial degeneration/necrosis and/or haemorrhage, cardiac atrioventricular valve hypertrophy/haemorrhage and atrial fibrovascular proliferation were seen in dogs (≥2 times human clinical exposure based on AUC). Focal arterial/perivascular inflammation in various tissues was observed in mice and an increased incidence of hepatic arterial degeneration and spontaneous cardiomyocyte degeneration with inflammation (spontaneous cardiomyopathy) was observed in rats (≥0.5 and 0.6 times human clinical exposure for rats and mice, respectively). Hepatic effects, including hepatocellular necrosis and inflammation, were observed in mice (≥0.6 times human clinical exposure). Bronchoalveolar inflammation of the lungs was observed in several dogs at ≥20 mg/kg/day (≥9 times human clinical exposure based on AUC) and was associated with shallow and/or laboured breathing.
Reversible haematological effects have been observed in dogs and rats given dabrafenib. In studies of up to 13 weeks, decreases in reticulocyte counts and/or red cell mass were observed in dogs and rats (≥10 and 1.4 times human clinical exposure, respectively).
In juvenile toxicity studies in rats, effects on growth (shorter long bone length), renal toxicity (tubular deposits, increased incidence of cortical cysts and tubular basophilia and reversible increases in urea and/or creatinine concentrations) and testicular toxicity (degeneration and tubular dilation) were observed (≥0.2 times human clinical exposure based on AUC).
Dabrafenib was phototoxic in an in vitro mouse fibroblast 3T3 Neutral Red Uptake (NRU) assay and in vivo at doses ≥100 mg/kg (>44 times human clinical exposure based on Cmax) in an oral phototoxicity study in hairless mice.
Combination with trametinib
In a study in dogs in which dabrafenib and trametinib were given in combination for 4 weeks, signs of gastrointestinal toxicity and decreased lymphoid cellularity of the thymus were observed at lower exposures than in dogs given trametinib alone. Otherwise, similar toxicities were observed as in comparable monotherapy studies.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.