FROVA Tablet Ref.[10818] Active ingredients: Frovatriptan
Source: FDA, National Drug Code (US) Revision Year: 2018
12.1. Mechanism of Action
Frovatriptan binds with high affinity to 5-HT1B/1D receptors. The therapeutic activity of FROVA is thought to be due to the agonist effects at the 5-HT1B/1D receptors on intracranial blood vessels (including the arterio-venous anastomoses) and sensory nerves of the trigeminal system which result in cranial vessel constriction and inhibition of pro-inflammatory neuropeptide release.
12.3. Pharmacokinetics
The pharmacokinetics of frovatriptan are similar in migraine patients and healthy subjects.
Absorption
Mean maximum blood concentrations (Cmax) in patients are achieved approximately 2 to 4 hours after administration of a single oral dose of frovatriptan 2.5 mg. The absolute bioavailability of an oral dose of frovatriptan 2.5 mg in healthy subjects is about 20% in males and 30% in females. Food has no significant effect on the bioavailability of frovatriptan, but delays tmax by one hour.
Distribution
Binding of frovatriptan to serum proteins is low (approximately 15%). Reversible binding to blood cells at equilibrium is approximately 60%, resulting in a blood: plasma ratio of about 2:1 in both males and females. The mean steady state volume of distribution of frovatriptan following intravenous administration of 0.8 mg is 4.2 L/kg in males and 3.0 L/kg in females.
Metabolism
In vitro, cytochrome P450 1A2 appears to be the principal enzyme involved in the metabolism of frovatriptan. Following administration of a single oral dose of radiolabeled frovatriptan 2.5 mg to healthy male and female subjects, 32% of the dose was recovered in urine and 62% in feces. Radiolabeled compounds excreted in urine were unchanged frovatriptan, hydroxylated frovatriptan, N-acetyl desmethyl frovatriptan, hydroxylated N-acetyl desmethyl frovatriptan and desmethyl frovatriptan, together with several other minor metabolites. Desmethyl frovatriptan has lower affinity for 5-HT1B/1D receptors compared to the parent compound. The N-acetyl desmethyl metabolite has no significant affinity for 5-HT receptors. The activity of the other metabolites is unknown.
Elimination
After an intravenous dose, mean clearance of frovatriptan was 220 and 130 mL/min in males and females, respectively. Renal clearance accounted for about 40% (82 mL/min) and 45% (60 mL/min) of total clearance in males and females, respectively. The mean terminal elimination half-life of frovatriptan in both males and females is approximately 26 hours.
Special Populations
Hepatic Impairment
The AUC of frovatriptan in patients with mild (Child-Pugh 5-6) to moderate (Child-Pugh 7-9) hepatic impairment was about twice that of young, healthy subjects, but within the range observed in healthy elderly subjects and was considerably lower than the values attained with higher doses of frovatriptan (up to 40 mg), which were not associated with any serious adverse effects. There is no clinical or pharmacokinetic experience with FROVA in patients with severe hepatic impairment.
Renal Impairment
The pharmacokinetics of frovatriptan following a single oral dose of 2.5 mg was not different in patients with renal impairment (5 males and 6 females, creatinine clearance 16-73 mL/min) compared to subjects with normal renal function.
Age
Mean AUC of frovatriptan was 1.5- to 2-fold higher in healthy elderly subjects (age 65 to 77 years) compared to those in healthy younger subjects (age 21 to 37 years). There was no difference in tmax or t1/2 between the two populations.
Sex
There was no difference in the mean terminal elimination half-life of frovatriptan in males and females. Bioavailability was higher, and systemic exposure to frovatriptan was approximately 2-fold greater, in females than males, irrespective of age.
Race
The effect of race on the pharmacokinetics of frovatriptan has not been examined.
Drug Interaction Studies
Frovatriptan is not an inhibitor of human monoamine oxidase (MAO) enzymes or cytochrome P450 (isozymes 1A2, 2C9, 2C19, 2D6, 2E1, 3A4) in vitro at concentrations up to 250- to 500-fold higher than the highest blood concentrations observed in man at a dose of 2.5 mg. No induction of drug metabolizing enzymes was observed following multiple dosing of frovatriptan to rats or on addition to human hepatocytes in vitro. Although no clinical trials have been performed, it is unlikely that frovatriptan will affect the metabolism of co-administered drugs metabolized by these mechanisms.
Oral Contraceptives
Retrospective analysis of pharmacokinetic data from females across trials indicated that the mean Cmax and AUC of frovatriptan are 30% higher in those subjects taking oral contraceptives compared to those not taking oral contraceptives.
Ergotamine
The AUC and Cmax of frovatriptan (2 × 2.5 mg dose) were reduced by approximately 25% when co-administered with ergotamine tartrate [see Contraindications (4), Drug Interactions (7.1)].
Propranolol
Propranolol increased the AUC of frovatriptan 2.5 mg in males by 60% and in females by 29%. The Cmax of frovatriptan was increased 23% in males and 16% in females in the presence of propranolol. The tmax as well as half-life of frovatriptan, though slightly longer in the females, were not affected by concomitant administration of propranolol.
Moclobemide
The pharmacokinetic profile of frovatriptan was unaffected when a single oral dose of frovatriptan 2.5 mg was administered to healthy female subjects receiving the MAO-A inhibitor, moclobemide, at an oral dose of 150 mg twice a day for 8 days.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of orally administered frovatriptan was evaluated in an 84-week study in mice (4, 13, and 40 mg/kg/day), a 104-week study in rats (8.5, 27, and 85 mg/kg/day), and a 26-week study in p53 ( + / - ) transgenic mice (20, 62.5, 200, and 400 mg/kg/day). Although a maximum tolerated dose was not achieved in the 84-week mouse study and in female rats, plasma exposures at the highest doses studied were higher than that achieved in humans at the MRHD of 7.5 mg/day. There were no increases in tumor incidence in the 84-week mouse study at doses producing plasma exposures (AUC) 140 times that in humans at the MRHD. In the rat study, there was a statistically significant increase in the incidence of pituitary adenomas in males only at 85 mg/kg/day, a dose associated with a plasma AUC 250 times that in humans at the MRHD. In the 26-week p53 ( + / - ) transgenic mouse study, the incidence of subcutaneous sarcomas was increased in females at doses of 200 and 400 mg/kg/day.
These sarcomas were associated with subcutaneously implanted animal identification transponders, and are not considered to be relevant to humans. There were no other increases in tumor incidence of any type in any dose group.
Mutagenesis
Frovatriptan was clastogenic in human lymphocyte cultures, in the absence of metabolic activation. In the bacterial reverse mutation assay (Ames test), frovatriptan produced an equivocal response in the absence of metabolic activation. Frovatriptan was negative in an in vitro mouse lymphoma tk assay and an in vivo mouse bone marrow micronucleus test.
Impairment of Fertility
Male and female rats were dosed orally with frovatriptan prior to and during mating and in females up to implantation, at doses of 100, 500, and 1000 mg/kg/day (equivalent to approximately 130, 650, and 1300 times the MRHD on a mg/m² basis). At all dose levels, there was an increase in the number of females that mated on the first day of pairing compared to control animals. This occurred in conjunction with a prolongation of the estrous cycle. In addition, females had a decreased mean number of corpora lutea, and consequently a lower number of live fetuses per litter, which suggested a partial impairment of ovulation. There were no other fertility-related effects.
14. Clinical Studies
The efficacy of FROVA in the acute treatment of migraine headaches was demonstrated in four randomized, double-blind, placebo-controlled, short-term outpatient trials. In these trials, patients received doses of frovatriptan from 0.5 mg to 40 mg. In these controlled short-term trials, patients were predominately female (88%) and Caucasian (94%) with a mean age of 42 years (range 18 to 69). Patients were instructed to treat a moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed for up to 24 hours after dosing. The associated symptoms nausea, vomiting, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours post dose. In two of the trials a second dose of FROVA was provided after the initial treatment, to treat recurrence of the headache within 24 hours. Other medication, excluding other 5-HT1 agonists and ergotamine containing compounds, was permitted from 2 hours after the first dose of FROVA. The frequency and time to use of additional medications were also recorded.
In all four placebo-controlled trials, the percentage of patients achieving a headache response 2 hours after treatment was significantly greater for those taking FROVA 2.5 mg compared to those taking placebo (Table 2).
Lower doses of frovatriptan (1 mg or 0.5 mg) were not effective at 2 hours. Higher doses (5 mg to 40 mg) of frovatriptan showed no added benefit over 2.5 mg but did cause a greater incidence of adverse events.
Table 2. Percentage of Patients with Headache Response (Mild or No Headache) 2 Hours Following Treatmenta:
| Trial | FROVA 2.5 mg | Placebo |
|---|---|---|
| 1 | 42%* (n=90) | 22% (n=91) |
| 2 | 39%* (n=187) | 21% (n=99) |
| 3 | 46%** (n=672) | 27% (n=347) |
| 4 | 37%** (n=438) | 23% (n=225) |
a ITT observed data, excludes patients who had missing data or were asleep.
* p<0.05.
** p<0.001 in comparison with placebo.
The estimated probability of achieving an initial headache response by 2 hours following treatment is depicted in Figure 1.
Figure 1. Estimated Probability of Achieving Initial Headache Response Within 2 Hours:
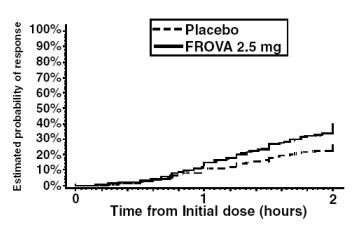
Figure 1 shows a Kaplan-Meier plot of the probability over time of obtaining headache response (no or mild pain) following treatment with FROVA 2.5 mg or placebo. The probabilities displayed are based on pooled data from the four placebo-controlled trials described in Table 2. Patients who did not achieve a response were censored at 24 hours.
In patients with migraine-associated nausea, photophobia, and phonophobia at baseline there was a decreased incidence of these symptoms in FROVA treated patients compared to placebo.
The estimated probability of patients taking a second dose or other medication for their migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
Figure 2. Estimated Probability of Patients Taking a Second Dose or Other Medication for Migraine Over the 24 Hours Following the Initial Dose of Study Treatment:

Figure 2 is a Kaplan-Meier plot showing the probability of patients taking a second dose or other medication for migraine over the 24 hours following the initial dose of study medication based on the data from the four placebo-controlled trials described in Table 2. The plot includes those patients who had a response to the initial dose and those who did not. The protocols did not permit remedication within 2 hours of the initial dose.
Efficacy was unaffected by a history of aura; gender; age, or concomitant medications commonly used by migraine patients.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.