HETRONIFLY Concentrate for solution for infusion Ref.[114653] Active ingredients: Serplulimab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Henlius Europe GmbH, Sternstraße 67, 40479 Düsseldorf, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, monoclonal antibodies and antibody drug conjugates, PD-1/PD-L1 (Programmed cell death-1/death ligand 1) inhibitors
ATC code: L01FF12
Mechanism of action
Serplulimab (HLX10) is a humanised monoclonal IgG4 antibody, which binds to the programmed cell death-1 (PD-1) receptor and blocks its interaction with ligands PD-L1 and PD-L2. The PD-1 receptor is a negative regulator of T-cell activity that has been shown to be involved in the control of T-cell immune responses. Engagement of PD-1 with the ligands PD-L1 and PD-L2, which are expressed in antigen presenting cells and may be expressed by tumours or other cells in the tumour microenvironment, results in inhibition of T-cell proliferation and cytokine secretion. Serplulimab potentiates T-cell responses, including anti-tumour responses, through blockade of PD-1 binding to PD-L1 and PD-L2 ligands.
The PD-1 receptor occupation of peripheral T cells and interleukin-2 (IL-2) release ability in vitro were studied in the phase 1 trial involving 29 Chinese patients with advanced solid tumour that were injected with single and multiple doses (0.3 mg/kg, 1 mg/kg, 3 mg/kg, 10 mg/kg) of serplulimab. The result showed that serplulimab could stably maintain the saturation state of receptor occupation and sustained functional blockage at the dosage from 0.3 mg/kg to 10 mg/kg every 2 weeks interval.
Clinical efficacy and safety
The efficacy of serplulimab in combination with chemotherapy (carboplatin plus etoposide) for the first-line treatment of ES-SCLC was evaluated in ASTRUM-005 trial (NCT04063163), a phase 3, randomised, double-blind, multiregional clinical trial. The primary efficacy endpoint was overall survival (OS). Secondary efficacy endpoints were progression free survival (PFS), objective response rate (ORR) and duration of response (DOR) as assessed by independent radiology review committee (IRRC) and investigator based on RECIST 1.1. Analysis for the primary endpoint was performed at 25 and 33 months since the start of the clinical trial. The study treatment regimens were unblinded after the primary analysis.
The trial included adult patients (18 years or older) with ES-SCLC (according to the Veterans Administration Lung Study Group [VALG] staging system) who had not been treated with systemic therapy and with an ECOG performance-status score of 0 or 1. Patients were excluded if they had active or untreated central nervous system metastases; active autoimmune disease; administration of systemic immunosuppressive medicinal products within 14 days prior to the first dose.
A total of 585 patients were enrolled and randomised (2:1) to receive one of the treatment regimens described in Table 3. Randomisation was stratified by PD-L1 expression level (negative: tumour proportion scores [TPS] <1%, positive: TPS ≥1%, or not evaluable/not available, measured by PD-L1 IHC 22C3 pharmDx kit), brain metastasis (yes versus no), and age (≥65 years versus <65 years).
Table 3. Intravenous treatment regimens:
| Treatment regimen | Induction (Four 21-Day Cycles) | Maintenance (21-Day Cycles) |
|---|---|---|
| A | Serplulimab (4.5 mg/kg) a + carboplatin (AUC=5, up to 750 mg)b + etoposide (100 mg/m²)b,c | Serplulimab (4.5 mg/kg)a |
| B | Placebo + carboplatin (AUC=5, up to 750 mg)b + etoposide (100 mg/m²)b,c | Placebo |
a Serplulimab was administered until disease progression or unacceptable toxicity.
b Carboplatin and etoposide were administered until completion of 4 cycles, or progressive disease or unacceptable toxicity, whichever occurred first.
c Etoposide was administered on day 1, 2 and 3 of each cycle.
Baseline characteristics were balanced between the treatment arms. Among the patients enrolled, 68.5% were Asian (401 patients), and 31.5% were non-Asian (184 patients), all of which were White. The median age was 62 years (range: 28-83) with 39.3% of patients ≥65 years of age, and 1.9% of patients ≥75 years of age. 82.2% of patients were men. Baseline ECOG performance-status score was 0 (17.6%) or 1 (82.4%). 16.9% of patients were PD-L1 positive (TPS ≥1%). 13.3% of patients had a history of brain metastases.
At the time of the interim analysis cut-off on 22 October 2021 when 66% of predefined OS events were observed (defined approximately 226, actual 246 OS events), patients had a median survival follow-up time of 12.3 months. OS, PFS and ORR results from the interim analysis are summarised in Table 4.
Table 4. Efficacy data at the primary analysis (data cut-off date: 22 October 2021):
| Arm A (Serplulimab + carboplatin + etoposide) | Arm B (Placebo + carboplatin + etoposide) | ||
|---|---|---|---|
| Number of patients | 389 | 196 | |
| Primary endpoint | |||
| OS | Number of patients with events, n (%) | 146 (37.5%) | 100 (51.0%) |
| Median OS (months) | 15.4 | 10.9 | |
| Hazard ratio (95% CI) | 0.63 (0.49-0.82) | ||
| p-value | <0.001 | ||
| Secondary endpoints | |||
| PFS -IRRC per RECIST 1.1 | Median PFS (months) | 5.7 | 4.3 |
| Hazard ratio (95% CI) | 0.48 (0.38-0.59) | ||
| Confirmed ORR | (%) | 67.4% | 58.7% |
| Median DOR | Months (95% CI) | 5.8 (5.2-7.5) | 4.1 (3.0-4.2) |
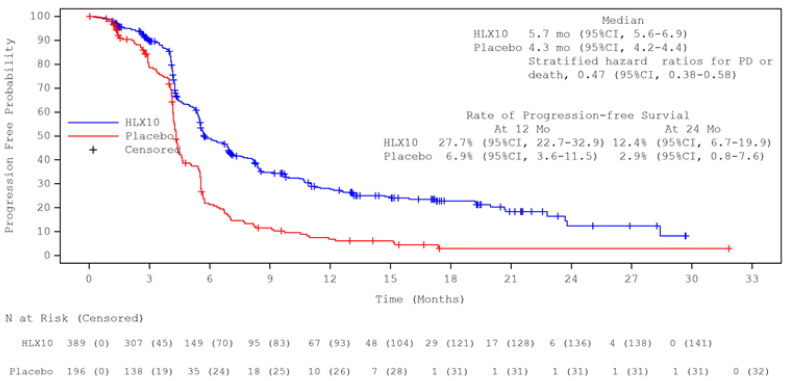
Updated analysis after unblinding with longer follow-up duration (median: 19.7 months) was conducted by the cut-off date 13 June 2022 when 100% of predefined OS events were observed (defined approximately 342, actual 363 OS events). The median OS was 15.8 months in the serplulimab group and 11.1 months in the placebo group. The stratified HR (95% CI) was 0.62 (0.50, 0.76). The median PFS by IRRC assessment per RECIST 1.1 was 5.7 months and 4.3 months, respectively, with a stratified HR (95% CI) of 0.47 (0.38, 0.58). The efficacy results of final analysis were consistent with the primary analysis. Kaplan-Meier curves for OS and PFS of final analysis are presented in Figures 1 and 2.
Figure 1. Kaplan-Meier curve of OS in overall population at the updated analysis (ITT) (data cut-off date: 13 June 2022):
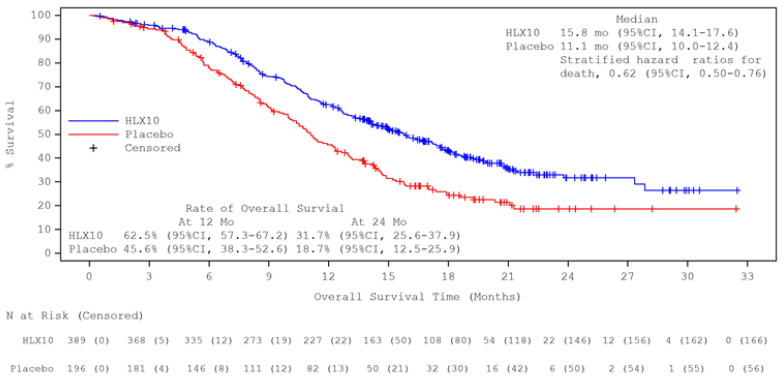
Figure 2. Kaplan-Meier curve of PFS (RECIST 1.1) by IRRC in overall population at the updated analysis (ITT) (data cut-off date: 13 June 2022):
Immunogenicity
The immunogenicity of serplulimab was evaluated in 389 patients treated with serplulimab at 4.5 mg/kg Q3W in the ASTRUM-005 trial. Seven patients (1.8%) were ADA positive at any visit, of whom 6 patients (1.5%) were treatment-emergent ADA positive, defined as at least one post-baseline ADA positive.
In dose escalation and dose expansion study HLX10-001, ADAs were observed in 13 out of 66 patients (19.7%).
Neutralising antibodies were not observed in either of the key studies. No evidence of ADA impact on pharmacokinetics, efficacy or safety was observed. However, data are still limited.
Elderly patients
In the ASTRUM-005 trial, of the 389 patients in the serplulimab group in the overall population, 153 (39.3%) were ≥65 years. No overall differences in efficacy were observed between elderly patients and younger patients.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with serplulimab in all subsets of the paediatric population for lung cancer (small cell and non-small cell lung cancer) (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Serplulimab pharmacokinetics has been investigated in a population pharmacokinetic (popPK) analysis that included 1 144 patients with lung cancer (including ES-SCLC) and other solid cancer types from 8 studies. The patients received serplulimab intravenously as monotherapy or combination therapy in the dose range of 0.3 to 10 mg/kg Q2W, 4.5 mg/kg Q3W, 200 mg Q2W, 300 mg Q3W and 400 mg Q4W. The PK was described by a two-compartment model with time-dependent clearance (CL). Inter-individual variability (coefficient of variation, CV) in base CL and central volume of distribution (Vc) was 25.8% and 15.4%. The mean (CV) observed trough concentration at steady state in the ASTRUM-005 trial was 62.5 μg/mL (36.3%).
Absorption
Serplulimab is administered by intravenous infusion and is therefore immediately and completely bioavailable. Other routes of administration have not been investigated.
Distribution
Based on a popPK analysis the volume of distribution of serplulimab is approximately 5.73 L.
Biotransformation
The metabolic pathway of serplulimab has not been characterised. Serplulimab is expected to be catabolised into small peptides and amino acids by general protein degradation processes.
Elimination
Based on a popPK analysis, serplulimab clearance (CL) after the first dose is 0.225 L/day. The clearance decreases over time by a maximum of 30.5% (CV 26.3%) with 106 days to reach half of the maximum effect. The half-life at steady state is approximately 24.3 days.
Linearity/non-linearity
Serplulimab exhibited linear pharmacokinetics over the dose range of 0.3 to 10 mg/kg Q2W (including flat doses of 200 mg Q2W, 300 mg Q3W and 400 mg Q4W) both after single and multiple doses.
Special populations
No dedicated studies have been performed in special populations. A popPK analysis suggested no difference in the total systemic clearance of serplulimab based on age (23-83 years), race (n=247 Whites and n=895 Asians), and ECOG performance-status score (0 or 1). Serplulimab clearance increased with increasing body weight.
Renal impairment
No effect of creatinine or creatinine clearance (CRCL) (Cockcroft-Gault) was found on serplulimab CL based on a popPK analysis in patients with mild (CRCL=60-89 ml/min; n=448), moderate (CRCL=30-59 ml/min; n=102), and severe (CRCL=15-29 ml/min; n=1) renal impairment, and normal renal function (CRCL≥ 90 ml/min, n=591). There are insufficient data in patients with severe renal impairment for dosing recommendations (see section 4.2).
Hepatic impairment
No effect of ALT, AST or total bilirubin was found on serplulimab CL based on a popPK analysis in patients with mild (BIL ≤ ULN and AST > ULN or BIL > 1 to 1.5 × ULN and any AST; n=176) and moderate (BIL > 1.5 to 3 × ULN and any AST; n=2) hepatic impairment, and normal (BIL ≤ ULN and AST ≤ ULN; n=956) hepatic function. There are insufficient data in patients with moderate hepatic impairment for dosing recommendations. Serplulimab has not been studied in patients with severe (BIL > 3 × ULN and any AST) hepatic impairment (see section 4.2).
5.3. Preclinical safety data
Repeat-dose toxicity
In the repeat-dose toxicity study in cynomolgus monkeys dosed for up to 31 weeks, a high incidence of pharmacology-related perivascular mononuclear cell infiltration in the brain choroid plexus was observed at 100 mg/kg. The no observed adverse effect level (NOAEL) in the 31-weeks toxicity study was 50 mg/kg/week, which produced exposure 36 times (calculated by AUC0-t) the exposure in humans at dose of 3 mg/kg every two weeks.
Reproductive toxicity
Reproductive toxicity studies have not been performed.
The PD-1/PD-L1 pathway is thought to be involved in maintaining tolerance to the foetus throughout pregnancy. Blockade of PD-L1 signalling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to result in an increase in foetal loss.
Two anti-PD-L1 monoclonal antibodies were evaluated in cynomolgus monkeys for reproductive and developmental toxicity and were shown to cause premature delivery, foetal loss and premature neonatal death when administrated to pregnant monkeys.
Therefore, potential risks of administering serplulimab during pregnancy include increased rates of abortion or stillbirth. Based on its mechanism of action, foetal exposure to serplulimab may increase the risk of developing immune-mediated disorders or altering the normal immune response and immune-related disorders that have been reported in PD-1 knockout mice.
Genotoxicity and carcinogenicity
No studies have been performed to assess the genotoxic or carcinogenic potential of serplulimab.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.