IMFINZI Concentrate for solution for infusion Ref.[8680] Active ingredients: Durvalumab
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies and antibody drug conjugates, PD-1/PDL-1 (Programmed cell death protein 1/death ligand 1) inhibitors
ATC code: L01FF03
Mechanism of action
Expression of programmed cell death ligand-1 (PD-L1) protein is an adaptive immune response that helps tumours evade detection and elimination by the immune system. PD-L1 can be induced by inflammatory signals (e.g., IFN-gamma) and can be expressed on both tumour cells and tumour-associated immune cells in tumour microenvironment. PD-L1 blocks T-cell function and activation through interaction with PD-1 and CD80 (B7.1). By binding to its receptors, PD-L1 reduces cytotoxic T-cell activity, proliferation and cytokine production.
Durvalumab is a fully human, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1). Durvalumab does not induce antibody dependent cell-mediated cytotoxicity (ADCC). Selective blockade of PD-L1/PD-1 and PD-L1/CD80 interactions enhances antitumour immune responses and increases T-cell activation.
The combination of tremelimumab, a CTLA-4 inhibitor and durvalumab, a PD-L1 inhibitor functions to enhance anti-tumour T-cell activation and function at multiple stages of the immune response resulting in improved anti-tumour responses. In murine syngeneic tumour models, dual blockade of PD-L1 and CTLA-4 resulted in enhanced anti-tumour activity.
Clinical efficacy and safety
Durvalumab doses of 10 mg/kg every 2 weeks, 1 120 mg every 3 weeks or 1 500 mg every 4 weeks were evaluated in NSCLC, ES-SCLC and endometrial cancer clinical studies. Based on the modeling and simulation of exposure, exposure-safety relationships and exposure-efficacy data comparisons, there are no anticipated clinically significant differences in efficacy and safety between durvalumab doses of 10 mg/kg every 2 weeks, 1 120 mg every 3 weeks or 1 500 mg every 4 weeks.
Resectable NSCLC – AEGEAN Study
AEGEAN was a randomised, double-blind, placebo-controlled, multicentre, Phase III study designed to evaluate the efficacy of IMFINZI in combination with platinum-based chemotherapy as neoadjuvant treatment, then continued as IMFINZI monotherapy after surgery, in patients with resectable NSCLC.
The following selection criteria define patients with high risk of recurrence who are included in the therapeutic indication and are reflective of a patient population with Stage IIA to select Stage IIIB as per the AJCC/UICC, 8th edition staging system:
- any patient with a tumour size ≥4 cm;
- any patient with N1 or N2 disease (regardless of primary tumour size), including multi-station N2 disease;
- patients with multiple tumour nodules in the same lobe or tumours that involve the main bronchus or tumours that invade visceral pleura, chest wall (including the parietal pleura and superior sulcus tumours), phrenic nerve or parietal pericardium; or tumours that are associated with atelectasis or obstructive pneumonitis that extends to the hilar region or involves part or all of the lung.
The study enrolled previously untreated patients with documented squamous or non-squamous NSCLC and no prior exposure to immune-mediated therapy, a WHO/ECOG Performance status of 0 or 1, and at least one RECIST 1.1 target lesion. Prior to randomisation, patients had tumour PD-L1 expression status confirmed using the VENTANA PD-L1 (SP263) Assay.
The study excluded patients with active or prior documented autoimmune disease, or use of immunosuppressive medication within 14 days of the first dose of durvalumab. The study population for efficacy analysis (modified intent-to-treat [mITT]) excluded patients with known EGFR mutations or ALK rearrangements. Following a protocol amendment, local ALK testing (unless squamous histology) and central EGFR testing was mandated. There were 51 patients with EGFR mutations and 11 patients with ALK rearrangements randomised into and treated within the study; however, these patients were not included in the mITT efficacy analysis and robust conclusions cannot be drawn regarding patients with EGFR mutations or ALK rearrangements.
Randomisation was stratified by disease stage (Stage II vs. Stage III) and by PD-L1 expression (TC <1% vs. TC ≥1%) status.
Post-operative radiotherapy (PORT) was permitted for patients for whom it was indicated according to local guidance. PORT was to be started within 8 weeks of surgery and adjuvant durvalumab/placebo must then have been started within 3 weeks of the completion of PORT.
The AEGEAN study randomised 802 patients in a 1:1 ratio to receive perioperative IMFINZI (Arm 1) or placebo (Arm 2) in combination with neoadjuvant chemotherapy. Crossover between the study arms was not permitted.
- Arm 1: IMFINZI 1 500 mg + chemotherapy every 3 weeks for up to 4 cycles prior to surgery, followed by IMFINZI 1 500 mg every 4 weeks for up to 12 cycles after surgery.
- Arm 2: Placebo + chemotherapy every 3 weeks for up to 4 cycles prior to surgery, followed by Placebo every 4 weeks for up to 12 cycles after surgery.
In the 2 treatment arms, patients received one of the following histology-based chemotherapy regimens:
- Squamous NSCLC
- Carboplatin + paclitaxel: carboplatin AUC 6 and paclitaxel 200 mg/m² via IV infusion on Day 1 of each 3-week cycle, for 4 cycles.
- Squamous NSCLC
- Cisplatin + gemcitabine: cisplatin 75 mg/m² via IV infusion on Day 1 of each 3-week cycle, for 4 cycles, and gemcitabine 1250 mg/m² via IV infusion on Day 1 and Day 8 of each 3-week cycle, for 4 cycles.
- Non-squamous NSCLC
- Pemetrexed + cisplatin: pemetrexed 500 mg/m² and cisplatin 75 mg/m² via IV infusion on Day 1 of each 3-week cycle, for 4 cycles.
- Non-squamous NSCLC
- Pemetrexed + carboplatin: pemetrexed 500 mg/m² and carboplatin AUC 5 via IV infusion on Day 1 of each 3-week cycle, for 4 cycles.
In the event of unfavourable tolerability, patients could switch from cisplatin to carboplatin therapy at any point and in patients with comorbidities or unable to tolerate cisplatin per Investigators judgement, carboplatin AUC 5 could be administered from cycle 1.
A RECIST 1.1 tumour assessment was performed at baseline, and upon completion of the neoadjuvant period (prior to surgery). The first post-surgical CT/MRI scan of the chest and abdomen (including the entire liver and both adrenals) was acquired 5 weeks ± 2 weeks after surgery and prior to, but as close as possible to the start of adjuvant therapy. Tumour assessments were then conducted every 12 weeks (relative to the date of surgery) until week 48, every 24 weeks (relative to the date of surgery) until week 192 (approximately 4 years), and then every 48 weeks (relative to the date of surgery) thereafter until RECIST 1.1 defined radiological PD, consent withdrawal, or death. Survival assessments were conducted at month 2, 3, and 4 following treatment discontinuation and then every 2 months until month 12 followed by every 3 months.
The primary endpoints of the study were pathological complete response (pCR) by blinded central pathology review, and event-free survival (EFS) by blinded independent central review (BICR) assessment. OS was a key secondary endpoint.
Efficacy analysis was conducted based on 740 patients in the mITT population: 366 patients in Arm 1 and 374 patients in Arm 2. Baseline demographics and disease characteristics of the population were as follows: male (71.6%), female (28.4%), age ≥ 65 years (51.6%), median age 65 years (range: 30 to 88), WHO/ECOG PS 0 (68.4%), WHO/ECOG PS 1 (31.6), White (53.6%), Asian (41.5%), Black or African American (0.9%), American Indian or Alaska Native (1.4%), Other Race (2.6%), Hispanic or Latino (16.1%), Not Hispanic or Latino (83.9%), current or past smokers (85.5%), never smoker (14.5%), squamous histology (48.6%) and non-squamous histology (50.7%), Stage II (28.4%), Stage III (71.6%), PD-L1 expression status TC ≥1% (66.6%), PD-L1 expression status TC <1% (33.4%).
In the mITT population, there were 295 (80.6%) patients in Arm 1 who underwent curative intent surgery compared to 302 (80.7%) patients in Arm 2. The number of patients who underwent PORT were 26 (7.1%) in Arm 1 and 24 (6.4%) in Arm 2.
At the primary (pre-specified) EFS analysis (DCO: 10 November 2022), with a maturity of 31.9% and a median EFS follow-up in censored patients of 11.7 months, the study showed a statistically significant improvement in the IMFINZI arm compared to the placebo arm [HR=0.68 (95% CI: 0.53, 0.88), p=0.003902].
At the updated (pre-specified) EFS analysis (DCO: 10 May 2024), the median EFS follow-up in censored patients was 25.9 months. At this analysis, OS was not formally tested for statistical significance; the HR for OS was 0.89 (95% CI: 0.70, 1.14) for IMFINZI arm compared to the placebo arm.
Table 5. Efficacy Results for the AEGEAN Study (mITT):
| IMFINZI + chemotherapy (N=366) | Placebo + chemotherapy (N=374) | |
|---|---|---|
| EFSa,c | ||
| Number of events, n (%) | 124 (33.9) | 165 (44.1) |
| Median EFS (95% CI) (months) | NR (42.3, NR) | 30 (20.6, NR) |
| Hazard ratio (95% CI) | 0.69 (0.55, 0.88) | |
| pCRa,b,c | ||
| Number of patients with response | 63 | 16 |
| Response rate, % (95% CI) | 17.21 (13.49, 21.48) | 4.28 (2.46, 6.85) |
| Difference in proportions, % (95% CI) | 12.96 (8.67, 17.57) | |
a Results are based on updated (pre-specified) EFS analysis (DCO: 10 May 2024) and pCR final analysis (DCO: 10 November 2022).
b Based on a pre-specified pCR interim analysis (DCO: 14 January 2022) in n=402, the pCR rate was statistically significant (p=0.000036) compared to significance level of 0.0082%.
c The 2-sided p-value for pCR was calculated based on a stratified CMH test. The 2-sided p-value for EFS was calculated based on a stratified log-rank test. Stratification factors included baseline PD-L1 and disease stage.
The boundary for declaring statistical significance for each of the efficacy endpoints were determined by a Lan-DeMets alpha spending function that approximates an O'Brien Fleming approach (EFS=0.9899%, pCR=0.0082%, 2-sided).
Figure 1. Kaplan-Meier Curve of updated EFS analysis (DCO: 10 May 2024):

NSCLC – PACIFIC Study
The efficacy of IMFINZI was evaluated in the PACIFIC Study, a randomised, double-blind, placebo-controlled, multicentre study in 713 patients with locally advanced, unresectable NSCLC. Patients had completed at least 2 cycles of definitive platinum-based chemotherapy with radiation therapy within 1 to 42 days prior to initiation of the study and had a ECOG performance status of 0 or 1. Ninety-two percent of patients had received a total dose of 54 to 66 Gy of radiation. The study excluded patients who had progressed following chemoradiation therapy, patients with prior exposure to any anti-PD-1 or anti-PD-L1 antibody, patients with active or prior documented autoimmune disease within 2 years of initiation of the study; a history of immunodeficiency; a history of severe immune-mediated adverse reactions; medical conditions that required systemic immunosuppression, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI. Patients were randomised 2:1 to receive 10 mg/kg IMFINZI (n=476) or 10 mg/kg placebo (n=237) via intravenous infusion every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed disease progression. Randomisation was stratified by gender, age (<65 years vs. ≥65 years) and smoking status (smoker vs. non-smoker). Patients with disease control at 12 months were given the option to be re-treated upon disease progression. Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter.
Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, archival tumour tissue specimens taken prior to chemoradiation therapy were retrospectively tested for PD-L1 expression on tumour cells (TC) using the VENTANA PD-L1 (SP263) IHC assay. Of the 713 patients randomised, 63% of patients provided a tissue sample of sufficient quality and quantity to determine PD-L1 expression and 37% were unknown.
The demographics and baseline disease characteristics were well balanced between study arms. Baseline demographics of the overall study population were as follows: male (70%), age ≥65 years (45%), age ≥75 years (8%), White (69%), Asian (27%), other (4%), current smoker (16%), past-smoker (75%), never smoker (9%), ECOG Performance Status 0 (49%), ECOG Performance Status 1 (51%). Disease characteristics were as follows: Stage IIIA (53%), Stage IIIB (45%), histological sub-groups of squamous (46%), non-squamous (54%). Of 451 patients with PD-L1 expression available, 67% were TC ≥1% [PD-L1 TC 1-24% (32%), PD-L1 TC ≥25% (35%)] and 33% were TC <1%.
The two primary endpoints of the study were progression-free survival (PFS) and overall survival (OS) of IMFINZI vs. placebo. Secondary efficacy endpoints included PFS at 12 months (PFS 12) and 18 months (PFS 18) from randomisation and Time from Randomisation to Second Progression (PFS2). PFS was assessed by Blinded Independent Central Review (BICR) according to RECIST v1.1.
The study demonstrated a statistically significant improvement in PFS in the IMFINZI-treated group compared with the placebo group [hazard ratio (HR)=0.52 (95% CI: 0.42, 0.65), p<0.0001]. The study demonstrated a statistically significant improvement in OS in the IMFINZI-treated group compared with the placebo group [HR=0.68 (95% CI: 0.53, 0.87), p=0.00251].
In the 5 year follow-up analysis, with a median follow-up of 34.2 months, IMFINZI continued to demonstrate improved OS and PFS compared to placebo. The OS and PFS results from the primary analysis and the follow-up analysis are summarized in Table 6.
Table 6. Efficacy results for the PACIFIC Study:
| Primary analysisa | 5 year follow-up analysisb | |||
|---|---|---|---|---|
| IMFINZI (n=476) | Placebo (n=237) | IMFINZI (n=476) | Placebo (n=237) | |
| OS | ||||
| Number of deaths (%) | 183 (38.4%) | 116 (48.9%) | 264 (55.5%) | 155 (65.4%) |
| Median (months) (95% CI) | NR (34.7, NR) | 28.7 (22.9, NR) | 47.5 (38.1, 52.9) | 29.1 (22.1, 35.1) |
| HR (95% CI) | 0.68 (0.53, 0.87) | 0.72 (0.59, 0.89) | ||
| 2-sided p-value | 0.00251 | |||
| OS at 24 months (%) (95% CI) | 66.3% (61.7%, 70.4%) | 55.6% (48.9%, 61.3%) | 66.3% (61.8%, 70.4%) | 55.3% (48.6%, 61.4%) |
| p-value | 0.005 | |||
| OS at 48 months (%) (95% CI) | 49.7% (45.0%, 54.2%) | 36.3% (30.1%, 42.6%) | ||
| OS at 60 months (%) (95% CI) | 42.9% (38.2%, 47.4%) | 33.4% (27.3%, 39.6%) | ||
| PFS | ||||
| Number of events (%) | 214 (45.0%) | 157 (66.2%) | 268 (56.3%) | 175 (73.8%) |
| Median PFS (months) (95% CI) | 16.8 (13.0, 18.1) | 5.6 (4.6, 7.8) | 16.9 (13.0, 23.9) | 5.6 (4.8, 7.7) |
| HR (95% CI) | 0.52 (0.42, 0.65) | 0.55 (0.45, 0.68) | ||
| p-value | p<0.0001 | |||
| PFS at 12 months (%) (95% CI) | 55.9% (51.0%, 60.4%) | 35.3% (29.0%, 41.7%) | 55.7% (51.0%, 60.2%) | 34.5% (28.3%, 40.8%) |
| PFS at 18 months (%) (95% CI) | 44.2% (37.7%, 50.5%) | 27.0% (19.9%, 34.5%) | 49.1% (44.2%, 53.8%) | 27.5% (21.6%, 33.6%) |
| PFS at 48 months (%) (95% CI) | 35.0% (29.9%, 40.1%) | 19.9% (14.4%, 26.1%) | ||
| PFS at 60 months (%) (95% CI) | 33.1% (28.0%, 38.2%) | 19.0% (13.6%, 25.2%) | ||
| PFS2c | ||||
| Median PFS2 (months) (95% CI) | 28.3 (25.1, 34.7) | 17.1 (14.5, 20.7) | ||
| HR (95% CI) | 0.58 (0.46, 0.73) | |||
| p-value | p<0.0001 | |||
a Primary analysis of PFS at data cut-off 13 February 2017. Primary analysis of OS and PFS2 at data cut-off 22 March 2018.
b Follow-up OS and PFS analysis at data cut-off 11 January 2021.
c PFS2 is defined as the time from the date of randomisation until the date of second progression (defined by local standard clinical practice) or death.
NR: Not Reached
Kaplan-Meier curves for OS and PFS from the 5 year follow-up analysis are presented in Figures 2 and 3.
Figure 2. Kaplan-Meier curve of OS:
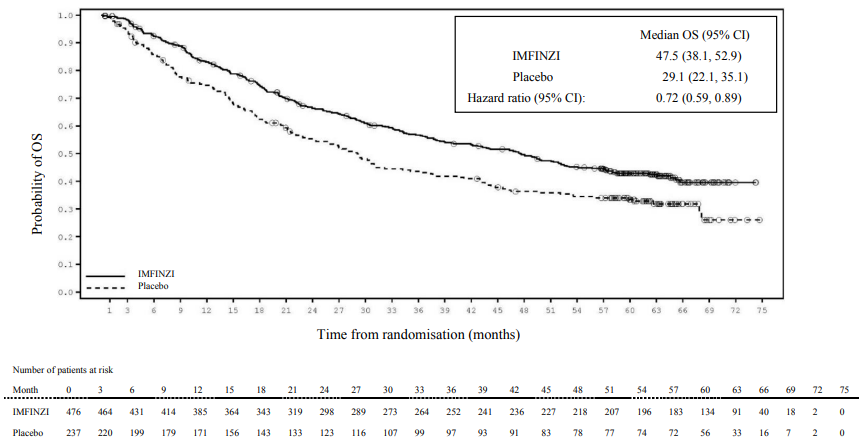
Figure 3. Kaplan-Meier curve of PFS:

The improvements in PFS and OS in favour of patients receiving IMFINZI compared to those receiving placebo were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history, EGFR mutation status and histology.
Post-hoc subgroup analysis by PD-L1 expression
Additional subgroup analyses were conducted to evaluate the efficacy by tumour PD-L1 expression (≥25%, 1-24%, ≥1%, <1%) and for patients whose PD-L1 status cannot be established (PD-L1 unknown). PFS and OS results from the 5 year follow-up analysis are summarised in Figures 4, 5, 6 and 7.
Figure 4. Kaplan-Meier curve of OS for PD-L1 TC ≥1%:
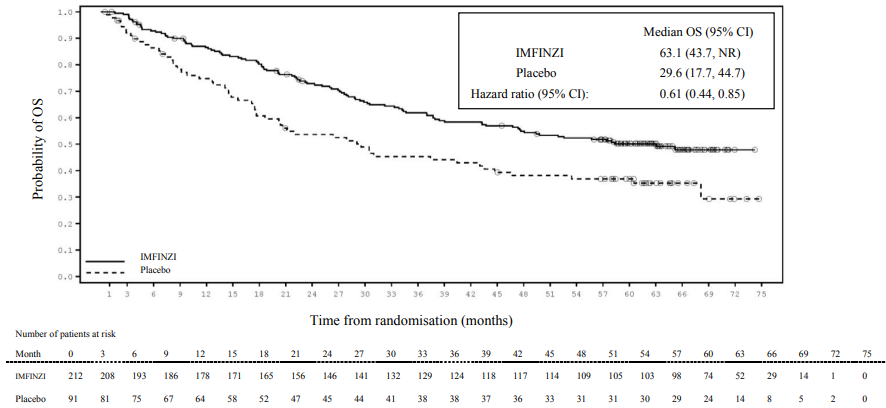
Figure 5. Kaplan-Meier curve of PFS for PD-L1 TC ≥1%:
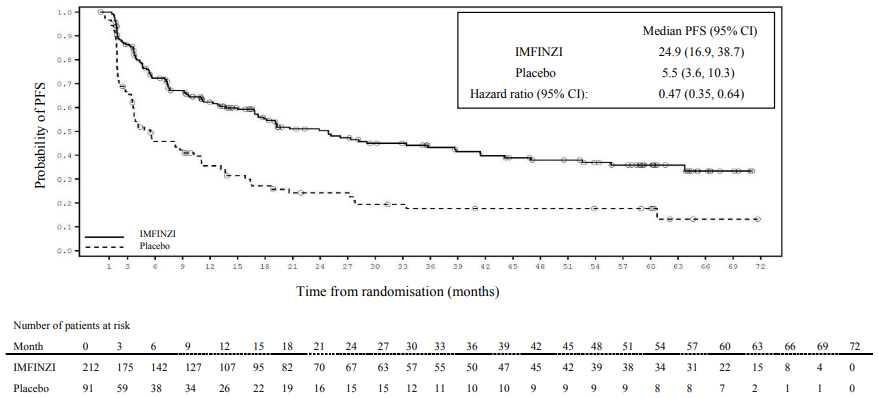
Figure 6. Forest plot of OS by PD-L1 expression:
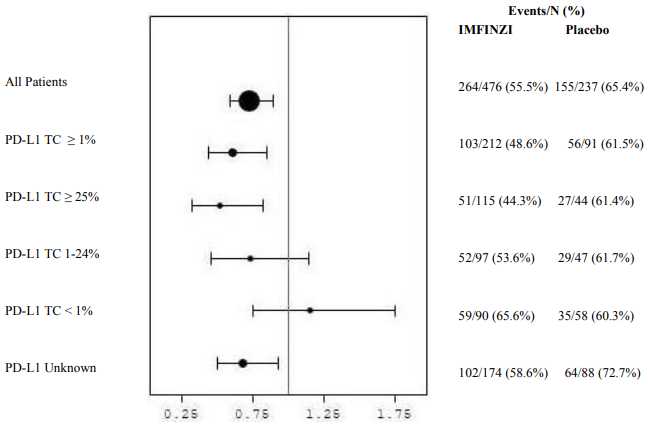
Figure 7. Forest plot of PFS by PD-L1 expression:
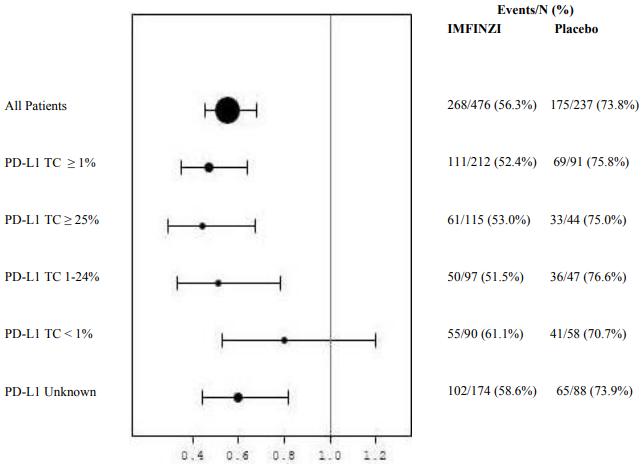
Overall the safety profile of durvalumab in PD-L1 TC ≥1% subgroup was consistent with the intent to treat population, as was the PD-L1 TC <1% subgroup.
Patient-reported outcomes (PRO)
Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 and C30 were assessed at baseline, every 4 weeks for the first 8 weeks, followed by every 8 weeks until completion of the treatment period or discontinuation of IMFINZI due to toxicity or disease progression. Compliance was similar between the IMFINZI and placebo treatment groups (83% vs. 85.1% overall of evaluable forms completed).
At baseline, no differences in patient-reported symptoms, function and HRQoL were observed between IMFINZI and placebo groups. Throughout the duration of the study to Week 48, there was no clinically meaningful difference between IMFINZI and placebo groups in symptoms, functioning and HRQoL (as assessed by a difference of greater than or equal to 10 points).
NSCLC – POSEIDON Study
POSEIDON was a study designed to evaluate the efficacy of IMFINZI with or without tremelimumab in combination with platinum-based chemotherapy. POSEIDON was a randomised, open-label, multicenter study in 1013 metastatic NSCLC patients with no sensitising epidermal growth factor receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) genomic tumour aberrations. Patients with histologically or cytologically documented metastatic NSCLC were eligible for enrolment. Patients had no prior chemotherapy or any other systemic therapy for metastatic NSCLC. Prior to randomisation, patients had tumour PD-L1 status confirmed by using the Ventana PD-L1 (SP263) assay. Patients had a World Health Organization (WHO)/Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 at enrolment.
The study excluded patients with active or prior documented autoimmune disease; active and/or untreated brain metastases; a history of immunodeficiency; administration of systemic immunosuppression within 14 days before the start of IMFINZI or tremelimumab, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI and/or tremelimumab (see section 4.4).
Randomisation was stratified by tumour cells (TC) PD-L1 expression (TC ≥50% vs. TC <50%), disease stage (Stage IVA vs. Stage IVB, per the 8th edition of American Joint Committee on Cancer), and histology (non-squamous vs. squamous).
Patients were randomised 1:1:1 to receive:
- Arm 1: IMFINZI 1 500 mg with tremelimumab 75 mg and platinum-based chemotherapy every 3 weeks for 4 cycles followed by, IMFINZI 1 500 mg every 4 weeks as monotherapy. A fifth dose of tremelimumab 75 mg was given at Week 16 alongside IMFINZI dose 6.
- Arm 2: IMFINZI 1 500 mg and platinum-based chemotherapy every 3 weeks for 4 cycles, followed by IMFINZI 1 500 mg every 4 weeks as monotherapy.
- Arm 3: Platinum-based chemotherapy every 3 weeks for 4 cycles. Patients could receive 2 additional cycles (a total of 6 cycles post-randomisation), as clinically indicated, at the Investigator's discretion.
In the 3 treatment arms, patients received one of the following histology-based chemotherapy regimens:
- Non-squamous NSCLC
- Pemetrexed 500 mg/m² with carboplatin AUC 5-6 or cisplatin 75 mg/m² every 3 weeks. Unless contraindicated by the investigator, pemetrexed maintenance could be given.
- Squamous NSCLC
- Gemcitabine 1 000 or 1 250 mg/m² on Days 1 and 8 with cisplatin 75 mg/m² or carboplatin AUC 5-6 on Day 1 every 3 weeks.
- Non-squamous or squamous NSCLC
- Nab-paclitaxel 100 mg/m² on Days 1, 8, and 15 with carboplatin AUC 5-6 on Day 1 every 3 weeks.
Tremelimumab was given up to a maximum of 5 doses unless there was disease progression or unacceptable toxicity. IMFINZI and histology-based pemetrexed maintenance therapy (when applicable) was continued until disease progression or unacceptable toxicity.
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The dual primary endpoints of the study were PFS and OS for IMFINZI + platinum-based chemotherapy vs. platinum-based chemotherapy alone. The key secondary endpoints of the study were PFS and OS for IMFINZI + tremelimumab + platinum-based chemotherapy and platinum-based chemotherapy alone. The secondary endpoints included objective response rate (ORR) and Duration of Response (DoR). PFS, ORR, and DoR, were assessed using BICR according to RECIST v1.1
The demographics and baseline disease characteristics were well-balanced between study arms. Baseline demographics of the overall study population were as follows: male (76.0%), age ≥65 years (47.1%), age ≥75 years (11.3%) median age 64 years (range: 27 to 87 years), White (55.9%), Asian (34.6%), Black or African American (2.0%), Other (7.6%), non-Hispanic or Latino (84.2%), current smoker or past-smoker (78.0%), WHO/ECOG PS 0 (33.4%), WHO/ECOG PS 1 (66.5%). Disease characteristics were as follows: Stage IVA (50.0%), Stage IVB (49.6%), histological sub-groups of squamous (36.9%), non-squamous (62.9%), brain metastases (10.5%), PD-L1 expression TC ≥50% (28.8%), PD-L1 expression TC <50% (71.1%).
The study showed a statistically significant improvement in OS with IMFINZI + tremelimumab + platinum-based chemotherapy vs. platinum-based chemotherapy. IMFINZI + tremelimumab + platinum-based chemotherapy showed a statistically significant improvement in PFS vs. platinum-based chemotherapy alone. The results are summarised below.
Table 7. Efficacy results for the POSEIDON study:
| Arm 1: IMFINZI+tremelimumab +platinum-based chemotherapy (n=338) | Arm 3: Platinum-based chemotherapy (n=337) | |
|---|---|---|
| OSa | ||
| Number of deaths (%) | 251 (74.3) | 285 (84.6) |
| Median OS (months) (95% CI) | 14.0 (11.7, 16.1) | 11.7 (10.5, 13.1) |
| HR (95% CI)b | 0.77 (0.650, 0.916) | |
| p-valuec | 0.00304 | |
| PFSa | ||
| Number of events (%) | 238 (70.4) | 258 (76.6) |
| Median PFS (months) (95% CI) | 6.2 (5.0, 6.5) | 4.8 (4.6, 5.8) |
| HR (95% CI)b | 0.72 (0.600, 0.860) | |
| p-valuec | 0.00031 | |
| ORR n (%) d,e | 130 (38.8) | 81 (24.4) |
| Complete Response n (%) | 2 (0.6) | 0 |
| Partial Response n (%) | 128 (38.2) | 81 (24.4) |
| Median DoR (months) (95% CI)d,e | 9.5 (7.2, NR) | 5.1 (4.4, 6.0) |
a Analysis of PFS at data cut off 24 July 2019 (median follow up 10.15 months). Analysis of OS at data cut off 12 March 2021 (median follow up 34.86 months). The boundaries for declaring efficacy (Arm 1 vs. Arm 3: PFS 0.00735, OS 0.00797; 2-sided) were determined by a Lan-DeMets alpha spending function that approximates an O'Brien Fleming approach. PFS was assessed by BICR according to RECIST v1.1.
b HR are derived using a Cox pH model stratified by PD-L1, histology and disease stage.
c 2-sided p-value based on a log-rank test stratified by PD-L1, histology and disease stage.
d Confirmed Objective Response.
e Post-hoc analysis.
NR = Not Reached, CI = Confidence Interval
Figure 8. Kaplan-Meier curve of OS:
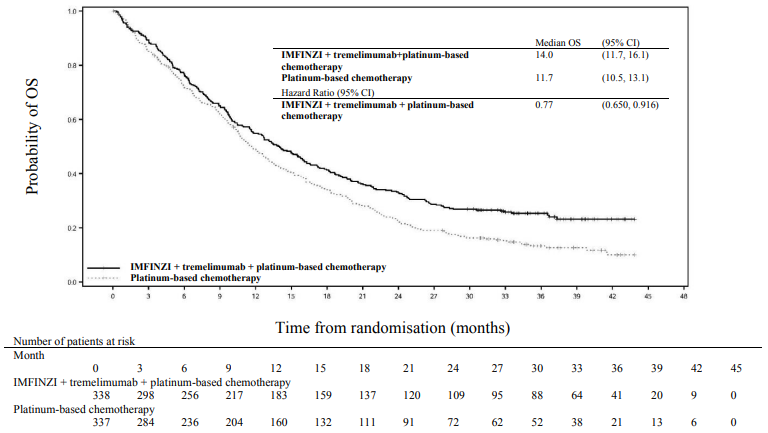
Figure 9. Kaplan-Meier curve of PFS:
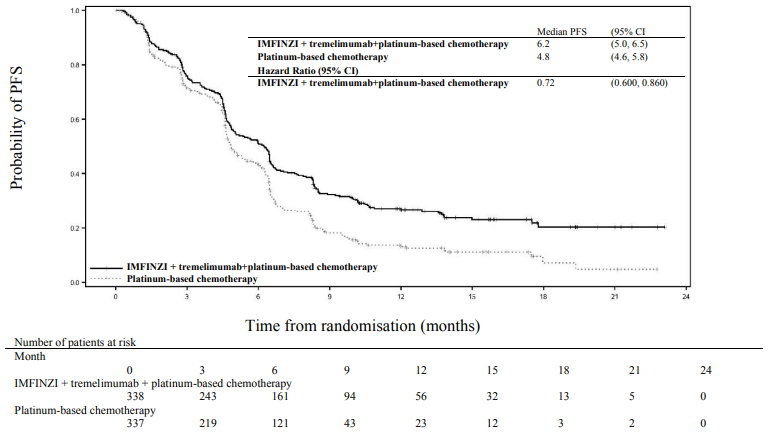
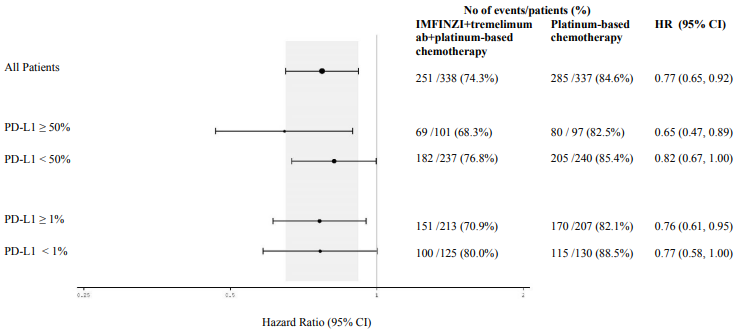
Figure 10 summarises efficacy results of OS by tumour PD-L1 expression in prespecified subgroup analyses.
Figure 10. Forest plot of OS by PD-L1 expression for IMFINZI+tremelimumab+platinum-based chemotherapy vs. platinum-based chemotherapy:
Elderly population
A total of 75 patients aged ≥75 years were enrolled in the IMFINZI in combination with tremelimumab and chemotherapy (n=35) and platinum-based chemotherapy only (n=40) arms of the POSEIDON study. An exploratory HR of 1.05 (95% CI: 0.64, 1.71) for OS was observed for the IMFINZI in combination with tremelimumab and platinum-based chemotherapy vs. platinum-based chemotherapy within this study subgroup. Due to the exploratory nature of this subgroup analysis no definitive conclusions can be drawn, but caution is suggested when considering this regimen for elderly patients.
SCLC – ADRIATIC Study
ADRIATIC was a study designed to evaluate the efficacy of IMFINZI with or without tremelimumab. ADRIATIC was a randomised, double-blind, placebo-controlled, multicentre study in 730 patients with histologically or cytologically confirmed LS-SCLC (Stage I to III according to AJCC, 8th edition) who had not progressed following concurrent chemoradiation therapy. Patients who were Stage I or II had to be medically inoperable as determined by the investigator. Patients completed 4 cycles of definitive platinum-based chemoradiation, 60-66 Gy once daily (QD) over 6 weeks or 45 Gy twice daily (BID) over 3 weeks, within 1 to 42 days prior to the first dose of study treatment. Prophylactic cranial irradiation (PCI) could be delivered at the discretion of the investigator after chemoradiation therapy and within 1 to 42 days prior to the first dose of study treatment. Patients had a WHO/ECOG performance status of 0 or 1 at enrolment.
The study excluded patients with active or prior documented autoimmune disease within 5 years of initiation of the study; a history of active primary immunodeficiency; a history of Grade ≥2 pneumonitis or active tuberculosis or hepatitis B or C or HIV infection and patients with active interstitial lung disease. Patients with mixed SCLC and NSCLC histology were also excluded.
Randomisation was stratified by stage (I/II vs. III) and receipt of PCI (yes vs. no). Patients were randomised 1:1:1 to receive:
- Arm 1: IMFINZI 1 500 mg + placebo every 4 weeks for 4 cycles, followed by IMFINZI 1 500 mg every 4 weeks.
- Arm 2: Placebo + a second placebo every 4 weeks for 4 cycles, followed by a single placebo every 4 weeks.
- Arm 3: IMFINZI 1 500 mg + tremelimumab 75 mg every 4 weeks for 4 cycles, followed by IMFINZI 1 500 mg every 4 weeks.
Once 600 patients had been randomised across all three arms, randomisation to arm 3 was complete and the subsequent 130 patients were randomised 1:1 to either arm 1 or 2, and received either IMFINZI 1 500 mg every 4 weeks or placebo every 4 weeks.
Treatment continued until disease progression, until unacceptable toxicity, or for a maximum of 24 months. Tumour assessments were conducted every 8 weeks for the first 72 weeks, then every 12 weeks up to 96 weeks and then every 24 weeks thereafter.
The demographics and baseline disease characteristics were well balanced between study arms. Baseline demographics and disease characteristics of the IMFINZI and placebo arms were as follows: male (69.1%), age ≥65 years (39.2%), White (50.4%), Black or African-American (0.8%), Asian (47.5%), other (1.3%), Hispanic or Latino (4.2%), current smoker (22.3%), past-smoker (68.5%), never smoker (9.2%), WHO/ECOG PS 0 (48.7%), WHO/ECOG PS 1 (51.3%), Stage I (3.6%), Stage II (9.1%), Stage III (87.4%).
Prior to randomisation, all patients received platinum-based chemotherapy (66.2% cisplatin-etoposide, 33.8% carboplatin-etoposide); 72.1% of patients received RT QD (of which 92.4% received ≥60 - ≤66 Gy QD); 27.9% received RT BID (of which 96.6% received 45 Gy BID) and 53.8% of patients received PCI. Response to CRT was as follows: complete response (12.3%), partial response (73.8%), stable disease (14.0%).
The dual primary endpoints of the study were OS and PFS of IMFINZI vs. placebo. Secondary efficacy endpoints included ORR of IMFINZI vs. placebo. PFS and ORR were assessed by BICR according to RECIST v1.1.
At a planned interim analysis, the study demonstrated a statistically significant improvement in OS and PFS for IMFINZI compared with placebo. See Table 8 and Figures 11 and 12.
Table 8. Efficacy results for the ADRIATIC study:
| Arm 1: IMFINZI (n=264) | Arm 2: Placebo (n=266) | |
|---|---|---|
| OSa | ||
| Number of deaths (%) | 115 (43.6) | 146 (54.9) |
| Median OS (months) (95% CI)b | 55.9 (37.3, NR) | 33.4 (25.5, 39.9) |
| HR (95% CI)c | 0.73 (0.569, 0.928) | |
| p-valued | 0.01042 | |
| PFSe | ||
| Number of events (%) | 139 (52.7) | 169 (63.5) |
| Median PFS (months) (95% CI)b | 16.6 (10.2, 28.2) | 9.2 (7.4, 12.9) |
| HR (95% CI)f | 0.76 (0.606, 0.950) | |
| p-valued | 0.01608 | |
a Median duration of OS follow-up in censored patients was 37.19 months in the IMFINZI arm and 37.24 months in the placebo arm.
b Calculated using the Kaplan Meier technique. CI for median derived based on Brookmeyer-Crowley method.
c The analysis for HR was performed using a stratified Cox proportional hazards model and the 2-sided p-value is based on a stratified log-rank test, both are adjusted for receipt of PCI.
d p-value based on the results from the pre-planned interim analysis. Based on a Lan-DeMets alpha spending function O'Brien Fleming type boundary and the actual number of events observed, the boundary for declaring statistical significance for OS was 0.01679 for a 4.5% overall alpha and for PFS was 0.02805 for a 5% overall alpha (Lan◦and◦DeMets 1983).
e Assessed by BICR according to RECIST v1.1.
f The analysis for HR was performed using a stratified Cox proportional hazards model and the 2-sided p-value is based on a stratified log-rank test, both are adjusted for TNM stage and receipt of PCI.
Figure 11. Kaplan-Meier Curve of OS:

Figure 12. Kaplan-Meier Curve of PFS:

SCLC – CASPIAN Study
CASPIAN was a study designed to evaluate the efficacy of IMFINZI with or without tremelimumab in combination with etoposide and either carboplatin or cisplatin. CASPIAN was a randomized, openlabel, multicentre study in 805 treatment naïve ES-SCLC patients with WHO/ECOG Performance status of 0 or 1, body weight >30 kg, suitable to receive a platinum-based chemotherapy regimen as first-line treatment for SCLC, with life expectancy ≥12 weeks, at least one target lesion by RECIST 1.1 and adequate organ and bone marrow function. Patients with asymptomatic or treated brain metastases were eligible. The study excluded patients with a history of chest radiation therapy; a history of active primary immunodeficiency; autoimmune disorders including paraneoplastic syndrome (PNS); active or prior documented autoimmune or inflammatory disorders; use of systemic immunosuppressants within 14 days before the first dose of the treatment except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI.
Randomisation was stratified by the planned platinum-based (carboplatin or cisplatin) therapy in cycle 1.
Patients were randomised 1:1:1 to receive:
- Arm 1: IMFINZI 1 500 mg + tremelimumab 75 mg + etoposide and either carboplatin or cisplatin.
- Arm 2: IMFINZI 1 500 mg + etoposide and either carboplatin or cisplatin.
- Arm 3: Either carboplatin (AUC 5 or 6 mg/ml/min) or cisplatin (75-80 mg/m²) on Day 1 and etoposide (80-100 mg/m²) intravenously on Days 1, 2, and 3 of each 21-day cycle for between 4–6 cycles.
For patients randomised to Arm 1 and 2, etoposide and either carboplatin or cisplatin was limited to 4 cycles on an every 3-week schedule subsequent to randomisation. IMFINZI monotherapy continued every 4 weeks until disease progression or unacceptable toxicity. Administration of IMFINZI monotherapy was permitted beyond disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator.
Patients randomised to Arm 3 were permitted to receive a total of up to 6 cycles of etoposide and either carboplatin or cisplatin. After completion of etoposide + platinum, PCI was permitted only in Arm 3 per investigator discretion.
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The primary endpoints of the study were OS of IMFINZI + etoposide + platinum (Arm 2) vs. etoposide + platinum alone (Arm 3) and IMFINZI + tremelimumab + etoposide + platinum (Arm 1) vs. etoposide + platinum alone (Arm 3). The key secondary endpoint was PFS. Other secondary endpoints were ORR, OS and PFS landmarks and PRO. PFS and ORR were assessed using Investigator assessments according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced between the two study arms (268 patients in Arm 2 and 269 patients in Arm 3). Baseline demographics of the overall study population were as follows: male (69.6%), age ≥ 65 years (39.6%), median age 63 years (range: 28 to 82 years), white (83.8%), Asian (14.5%), Black or African American (0.9%), other (0.6%), non-Hispanic or Latino (96.1%), current or past-smoker (93.1%), never smoker (6.9%), WHO/ECOG PS 0 (35.2%), WHO/ECOG PS 1 (64.8%), Stage IV 90.3%, 24.6% of the patients received cisplatin and 74.1% of the patients received carboplatin. In Arm 3, 56.8% of the patients received 6 cycles of etoposide + platinum and 7.8% of the patients received PCI.
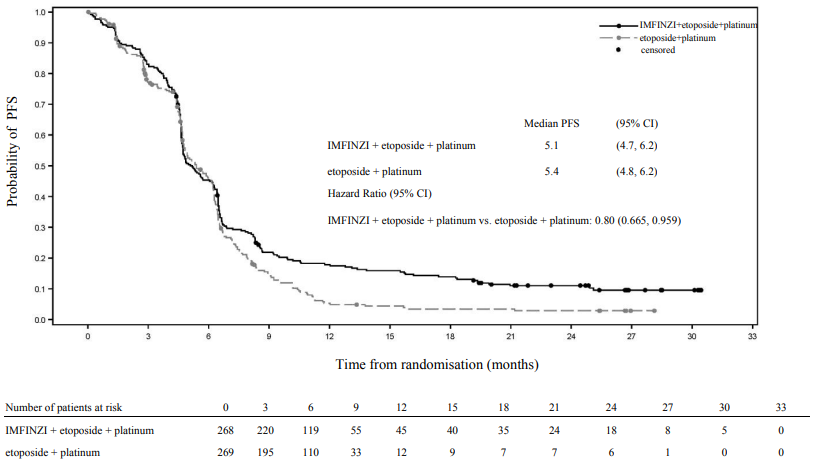
At a planned interim (primary) analysis the study demonstrated a statistically significant improvement in OS with IMFINZI + etoposide + platinum (Arm 2) vs. etoposide + platinum alone (Arm 3) [HR=0.73 (95% CI: 0.591, 0.909), p=0.0047]. Although not formally tested for significance, IMFINZI + etoposide + platinum demonstrated an improvement in PFS vs. etoposide + platinum alone [HR=0.78 (95% CI: 0.645, 0.936)].
The PFS, ORR and DoR results from the planned final analysis (DCO: 27 Jan 2020) are summarized in Table 9. Kaplan-Meier curve for PFS is presented in Figure 14.
The OS results with the planned long-term OS follow-up analysis (DCO: 22 March 2021) (median follow-up: 39.3 months) are presented in Table 9. IMFINZI + etoposide + platinum (Arm 2) vs. etoposide + platinum (Arm 3) continued to demonstrate sustained improvement in OS. Kaplan-Meier curve for OS is presented in Figure 13.
Table 9. Efficacy Results for the CASPIAN Study:
| Final analysisa | Long-term follow-up analysisb | |||
|---|---|---|---|---|
| Arm 2: IMFINZI + etoposide and either carboplatin or cisplatin (n=268) | Arm 3: etoposide + and either carboplatin or cisplatin (n=269) | Arm 2: IMFINZI + etoposide and either carboplatin or cisplatin (n=268) | Arm 3: etoposide + and either carboplatin or cisplatin (n=269) | |
| OS | ||||
| Number of deaths (%) | 210 (78.4) | 231 (85.9) | 221 (82.5) | 248 (92.2) |
| Median OS (months) (95% CI) | 12.9 (11.3, 14.7) | 10.5 (9.3, 11.2) | 12.9 (11.3, 14.7) | 10.5 (9.3, 11.2) |
| HR (95% CI)b,c | 0.75 (0.625, 0.910) | 0.71 (0.595, 0.858) | ||
| p-valued | 0.0032 | 0.0003 | ||
| OS at 18 months (%) (95% CI) | 32.0 (26.5, 37.7) | 24.8 (19.7, 30.1) | 32.0 (26.5, 37.7) | 24.8 (19.7, 30.1) |
| OS at 36 months (%) (95% CI) | 17.6 (13.3, 22.4) | 5.8 (3.4, 9.1) | ||
| PFS | ||||
| Number of events (%) | 234 (87.3) | 236 (87.7) | ||
| Median PFS (months) (95% CI) | 5.1 (4.7, 6.2) | 5.4 (4.8, 6.2) | ||
| HR (95% CI)c | 0.80 (0.665, 0.959) | |||
| PFS at 6 months (%) (95% CI) | 45.4 (39.3, 51.3) | 45.8 (39.5, 51.9) | ||
| PFS at 12 months (%) (95% CI) | 17.9 (13.5, 22.8) | 5.3 (2.9, 8.8) | ||
| ORR n (%) (95% CI)e | 182 (67.9) (62.0, 73.5) | 156 (58.0) (51.8, 64.0) | ||
| Complete Response n (%) | 7 (2.6) | 2 (0.7) | ||
| Partial Response n (%) | 175 (65.3) | 154 (57.2) | ||
| Median DoR (months) (95% CI)e,f | 5.1 (4.9, 5.3) | 5.1 (4.8, 5.3) | ||
a Final PFS, ORR and DoR analysis at data cut-off 27 January 2020.
b Long-term follow-up OS analysis at data cut-off 22 March 2021.
c The analysis was performed using the stratified log-rank test, adjusting for planned platinum therapy in Cycle 1 (carboplatin or cisplatin), and using the rank tests of association approach.
d At the interim analysis (data cut-off 11 March 2019) the OS p-value was 0.0047, which met the boundary for declaring statistical significance of 0.0178 for a 4% overall 2-sided alpha, based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary with the actual number of events observed.
e Confirmed Objective Response.
f Post-hoc analysis.
Figure 13. Kaplan-Meier curve of OS:

Figure 14. Kaplan-Meier curve of PFS:
Subgroup analysis
The improvements in OS in favour of patients receiving IMFINZI + etoposide + platinum compared to those receiving etoposide + platinum alone, were consistently observed across the prespecified subgroups based on demographics, geographical region, carboplatin or cisplatin use and disease characteristics.
BTC – TOPAZ-1 Study
TOPAZ-1 was a study designed to evaluate the efficacy of IMFINZI in combination with gemcitabine and cisplatin. TOPAZ-1 was a randomised, double-blind, placebo-controlled, multicentre study in 685 patients with unresectable or metastatic BTC (including intrahepatic and extrahepatic cholangiocarcinoma and gallbladder carcinoma) and ECOG Performance status of 0 or 1. Patients had not received previous therapy in the advanced/unresectable setting. Patients who developed recurrent disease >6 months after surgery and/or completion of adjuvant therapy were included. Patients must have had an adequate organ and bone marrow function, and have had acceptable serum bilirubin levels (≤2.0 x the upper limit of normal (ULN)), and any clinically significant biliary obstruction had to be resolved before randomisation.
The study excluded patients with ampullary carcinoma, with brain metastases, active or prior documented autoimmune or inflammatory disorders, HIV infection or active infections, including tuberculosis or hepatitis C or patients with current or prior use of immunosuppressive medication within 14 days before the first dose of IMFINZI. Patients with active HBV were allowed to participate if they were on antiviral therapy.
Randomisation was stratified by disease status (initially unresectable vs. recurrent) and primary tumour location (intrahepatic cholangiocarcinoma vs. extrahepatic cholangiocarcinoma vs. gallbladder carcinoma).
Patients were randomised 1:1 to receive:
- Arm 1: IMFINZI 1 500 mg administered on Day 1 + gemcitabine 1 000 mg/m² and cisplatin 25 mg/m² (each administered on Days 1 and 8) every 3 weeks (21 days) for up to 8 cycles, followed by IMFINZI 1 500 mg every 4 weeks until disease progression or unacceptable toxicity, or
- Arm 2: Placebo administered on Day 1 + gemcitabine 1 000 mg/m² and cisplatin 25 mg/m² (each administered on Days 1 and 8) every 3 weeks (21 days) for up to 8 cycles, followed by placebo every 4 weeks until disease progression or unacceptable toxicity.
Tumour assessments were conducted every 6 weeks for the first 24 weeks after the date of randomisation, and then every 8 weeks until confirmed objective disease progression.
The primary endpoint of the study was OS, the key secondary endpoint was PFS. Other secondary endpoints were ORR, DoR and PRO. PFS, ORR and DoR were investigator-assessed according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced between the two study arms (341 patients in Arm 1 and 344 patients in Arm 2). Baseline demographics of the overall study population were as follows: male (50.4%), age <65 years (53.3%), white (37.2%), Asian (56.4%), Black or African American (2.0%), other (4.2%), non-Hispanic or Latino (93.1%), ECOG PS 0 (49.1%), vs. PS 1 (50.9%), primary tumour location (intrahepatic bile duct 55.9%, extrahepatic bile duct 19.1% and gallbladder 25.0%), disease status [recurrent (19.1%) vs. unresectable (80.7%), metastatic (86.0%) vs. locally advanced (13.9%)]. PD-L1 expression was evaluated on tumour and immune cells using the Ventana PD-L1 (SP263) assay and the TAP (tumour area positivity) algorithm, 58.7% patients had TAP ≥1% and 30.1% TAP <1%.
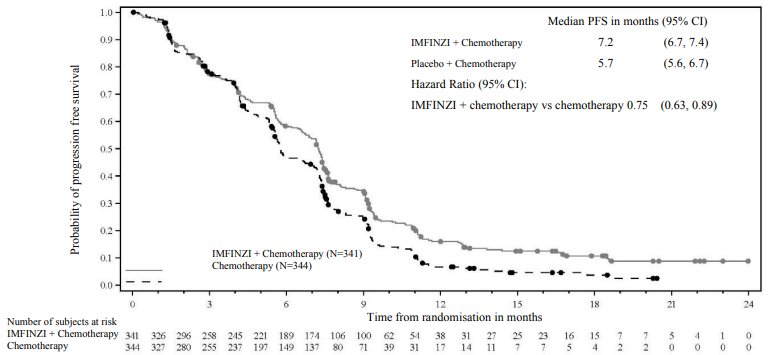
OS and PFS were formally tested at a pre-planned interim analysis (data cut-off 11 Aug 2021) after a median follow-up of 9.8 months. Efficacy results are shown in Table 10 and Figure 16. The maturity for OS was 62% and the maturity for PFS was 84%. IMFINZI + chemotherapy (Arm 1) showed statistically significant improvement vs. placebo + chemotherapy (Arm 2) in OS and in PFS.
Table 10. Efficacy Results for the TOPAZ-1 Studya:
| IMFINZI + gemcitabine and cisplatin (n=341) | Placebo + gemcitabine and cisplatin (n=344) | |
|---|---|---|
| OS | ||
| Number of deaths (%) | 198 (58.1) | 226 (65.7) |
| Median OS (months) (95% CI)b | 12.8 (11.1, 14.0) | 11.5 (10.1, 12.5) |
| HR (95% CI)c | 0.80 (0.66, 0.97) | |
| p-valuec,d | 0.021 | |
| Median follow-up in all patients (months) | 10.2 | 9.5 |
| PFS | ||
| Number of events (%) | 276 (80.9) | 297 (86.3) |
| Median PFS (months) (95% CI)b | 7.2 (6.7, 7.4) | 5.7 (5.6, 6.7) |
| HR (95% CI)c | 0.75 (0.63, 0.89) | |
| p-valuec,e | 0.001 | |
| Median follow-up in all patients (months) | 7.2 | 5.6 |
| ORRf | 91 (26.7) | 64 (18.7) |
| Complete Response n (%) | 7 (2.1) | 2 (0.6) |
| Partial Response n (%) | 84 (24.6) | 62 (18.1) |
| DoR | ||
| Median DoR (months) (95% CI)b | 6.4 (5.9, 8.1) | 6.2 (4.4, 7.3) |
a Analysis at data cut-off 11 August 2021.
b Calculated using the Kaplan-Meier technique. CI for median derived based on Brookmeyer-Crowley method.
c The analysis for HR was performed using a stratified Cox proportional hazards model and 2-sided p-value is based on a stratified log-rank test, both are adjusted for disease status and primary tumour location.
d At the interim analysis (data cut-off 11 August 2021) the OS p-value was 0.021, which met the boundary for declaring statistical significance of 0.03 for a 4.9% overall 2-sided alpha, based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary with the actual number of events observed.
e At the interim analysis (data cut-off 11 August 2021) the PFS p-value was 0.001, which met the boundary for declaring statistical significance of 0.0481 for a 4.9% overall 2-sided alpha, based on a Lan-DeMets alpha spending function with Pocock-type boundary with the actual number of events observed.
f Confirmed objective response.
An additional planned follow-up analysis of OS (data cut-off 25 Feb 2022) was performed 6.5 months after the interim analysis with an OS maturity of 77%. IMFINZI + chemotherapy continued to demonstrate improved OS vs. chemotherapy alone [HR=0.76, (95% CI: 0.64, 0.91)] and the median follow-up increased to 12 months.
Figure 15. Kaplan-Meier curve of OS, follow-up OS analysis at data cut-off 25 February 2022:
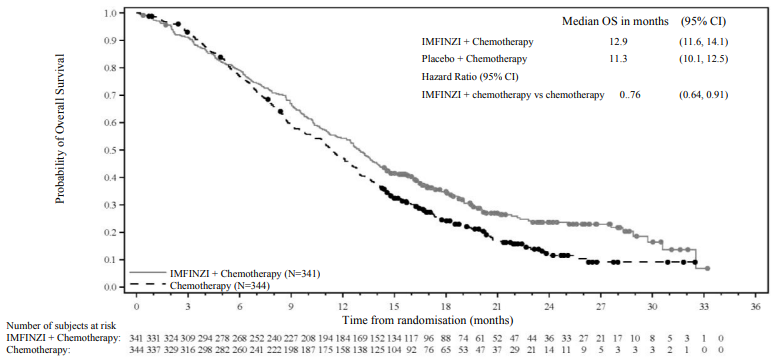
Figure 16. Kaplan-Meier curve of PFS, inferential (primary) analysis at data cut-off 11 August 2021:
HCC - HIMALAYA Study
The efficacy of IMFINZI as monotherapy and given in combination with a single dose of tremelimumab 300 mg was evaluated in the HIMALAYA Study, a randomised, open-label, multicentre study in patients with confirmed uHCC who did not receive prior systemic treatment for HCC. The study included patients with Barcelona Clinic Liver Cancer (BCLC) Stage C or B (not eligible for locoregional therapy) and Child-Pugh Score Class A.
The study excluded patients with brain metastases or a history of brain metastases, co-infection of viral hepatitis B and hepatitis C; active or prior documented gastrointestinal (GI) bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Patients with oesophageal varices were included except those with active or prior documented GI bleeding within 12 months prior to study entry.
Randomisation was stratified by macrovascular invasion (MVI) (yes vs. no), aetiology of liver disease (confirmed hepatitis B virus vs. confirmed hepatitis C virus vs. others) and ECOG performance status (0 vs. 1). The HIMALAYA study randomized 1171 patients 1:1:1 to receive:
- IMFINZI: durvalumab 1 500 mg every 4 weeks.
- Tremelimumab 300 mg as a single dose + IMFINZI 1 500 mg; followed by IMFINZI 1 500 mg every 4 weeks.
- Sorafenib 400 mg twice daily.
Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter. Survival assessments were conducted every month for the first 3 months following treatment discontinuation and then every 2 months.
The primary endpoint was OS superiority for the comparison of IMFINZI given in combination with a single dose of tremelimumab vs. Sorafenib. The key secondary objectives were OS non-inferiority followed by superiority for the comparison of IMFINZI vs. Sorafenib. Other secondary endpoints included PFS, Investigator-assessed ORR and DoR according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced among study arms. The baseline demographics of the overall study population were as follows: male (83.7%), age <65 years (50.4%) White (44.6%), Asian (50.7%), Black or African American (1.7%), Other race (2.3%), ECOG PS 0 (62.6%); Child-Pugh Class score A (99.5%), macrovascular invasion (25.2%), extrahepatic spread (53.4%), baseline AFP <400 ng/ml (63.7%), baseline AFP ≥400 ng/ml (34.5%), viral aetiology; hepatitis B (30.6%), hepatitis C (27.2%), uninfected (42.2%), evaluable PD-L1 data (86.3%), PD-L1 Tumour area positivity (TAP) ≥1% (38.9%), PD-L1 TAP <1% (48.3%) [Ventana PD-L1 (SP263) assay].
Results are presented in Table 11, Figure 17 and Figure 18.
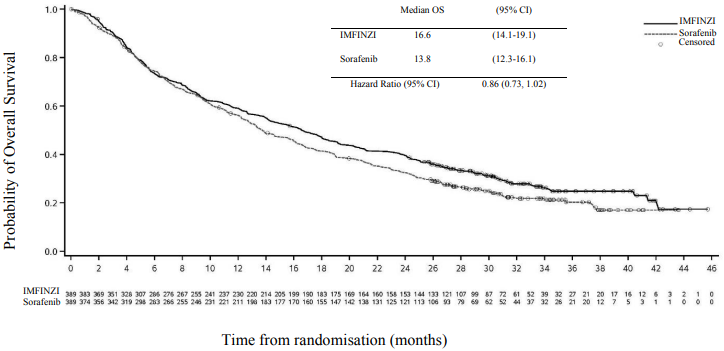
Table 11. Efficacy Results for the HIMALAYA Study for IMFINZI given in combination with a single dose of tremelimumab 300 mg and IMFINZI as monotherapy vs. Sorafenib:
| IMFINZI + tremelimumab 300 mg (n=393) | Sorafenib (n=389) | IMFINZI (n=389) | |
|---|---|---|---|
| Follow up duration | |||
| Median follow up (months)a | 33.2 | 32.2 | 32.6 |
| OS | |||
| Number of deaths (%) | 262 (66.7) | 293 (75.3) | 280 (72.0) |
| Median OS (months) (95% CI) | 16.4 (14.2, 19.6) | 13.8 (12.3, 16.1) | 16.6 (14.1, 19.1) |
| HR (95% CI)b,c | 0.78 (0.66, 0.92) | - | |
| p-valued | 0.0035 | - | |
| HR (95% CI)b,c,e | - | 0.86 (0.73, 1.03) | |
| PFS | |||
| Number of events (%) | 335 (85.2) | 327 (84.1) | 345 (88.7) |
| Median PFS (months) (95% CI) | 3.78 (3.68-5.32) | 4.07 (3.75-5.49) | 3.65 (3.19-3.75) |
| HR (95% CI) | 0.90 (0.77, 1.05) | - | |
| HR (95% CI) | - | 1.02 (0.88, 1.19) | |
| ORR | |||
| ORR n (%) f | 79 (20.1) | 20 (5.1) | 66 (17.0) |
| Complete Response n (%) | 12 (3.1) | 0 | 6 (1.5) |
| Partial Response n (%) | 67 (17.0) | 20 (5.1) | 60 (15.4) |
| DoR | |||
| Median DoR (months) | 22.3 | 18.4 | 16.8 |
a Calculated using reverse the Kaplan-Meier technique (with censor indicator reversed).
b Based on stratified Cox-model adjusting for treatment, etiology of liver disease (HBV versus HCV versus others), ECOG (0 versus 1).
c Performed using stratified log-rank test adjusting for treatment, etiology of liver disease (HBV versus HCV versus others), ECOG (0 versus 1), and macro-vascular invasion (yes versus no).
d Based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary and the actual number of events observed, the boundary for declaring statistical significance for IMFINZI + tremelimumab 300 mg vs. Sorafenib was 0.0398 (Lan◦and◦DeMets 1983).
e Non-inferiority margin for HR (IMFINZI vs Sorafenib) is 1.08 using a 95.67% confidence interval based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary and the actual number of events observed (Lan◦and◦DeMets 1983). P-value based on superiority testing of IMFINZI vs. Sorafenib was 0.0674 and did not reach statistical significance.
f Confirmed complete response.
CI=Confidence Interval
Figure 17. Kaplan-Meier curve of OS of IMFINI given in combination with a single dose of tremelimumab 300 mg:
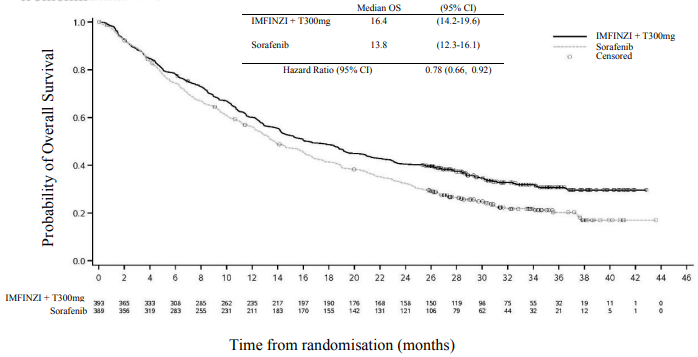
Figure 18. Kaplan-Meier curve of OS of IMFINZI given as monotherapy:
Endometrial Cancer – DUO-E Study
DUO-E was a randomised, multicentre, double-blind, placebo-controlled Phase III study of first-line platinum-based chemotherapy in combination with IMFINZI, followed by IMFINZI with or without olaparib in patients with advanced or recurrent endometrial cancer. Patients had to have endometrial cancer in one of the following categories: newly diagnosed Stage III disease (measurable disease per RECIST v1.1 following surgery or diagnostic biopsy), newly diagnosed Stage IV disease (with or without disease following surgery or diagnostic biopsy), or recurrence of disease (measurable or non-measurable disease per RECIST v1.1) where the potential for cure by surgery alone or in combination is poor. For patients with recurrent disease, prior chemotherapy was allowed only if it was administered in the adjuvant setting and there was at least 12 months from the date of last dose of chemotherapy administered to the date of subsequent relapse. The study included patients with epithelial endometrial carcinomas of all histologies, including carcinosarcomas. Patients with endometrial sarcoma were excluded.
Randomisation was stratified by tumour tissue's mismatch repair (MMR) status (proficient versus deficient), disease status (recurrent versus newly diagnosed), and geographic region (Asia versus rest of the world). Patients were randomised 1:1:1 to one of the following arms:
- Arm 1 (Platinum-based chemotherapy): Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with durvalumab placebo every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received durvalumab placebo every 4 weeks and olaparib placebo tablets twice daily as maintenance treatment until disease progression.
- Arm 2 (Platinum-based chemotherapy + IMFINZI): Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with 1 120 mg durvalumab every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received 1 500 mg durvalumab every 4 weeks with olaparib placebo tablets twice daily as maintenance treatment until disease progression.
- Arm 3 (Platinum-based chemotherapy + IMFINZI + olaparib): Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with 1 120 mg durvalumab every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received 1 500 mg durvalumab every 4 weeks with 300 mg olaparib tablets twice daily as maintenance treatment until disease progression.
Patients who discontinued either product (IMFINZI/placebo or olaparib/placebo) for reasons other than disease progression could continue treatment with the other product if appropriate based on toxicity considerations and investigator discretion.
Treatment was continued until RECIST v1.1-defined progression of disease or unacceptable toxicity. Assessment of tumour status was performed every 9 weeks for the first 18 weeks relative to randomisation and every 12 weeks thereafter.
The primary endpoint was PFS, determined by investigator assessment using RECIST v1.1. Secondary efficacy endpoints included OS, ORR and DoR.
The study demonstrated a statistically significant improvement in PFS in the ITT population, for patients treated with platinum-based chemotherapy + IMFINZI + olaparib compared to platinum-based chemotherapy [HR=0.55 (95% CI: 0.43, 0.69), p=<0.0001], and for patients treated with platinum-based chemotherapy + IMFINZI compared to platinum-based chemotherapy [HR=0.71 (95% CI: 0.57, 0.89), p=0.003]. At the time of PFS analysis, interim OS data were 28% mature with events in 199 of 718 patients.
Mismatch repair (MMR) status was determined centrally using an MMR immunohistochemistry panel assay. Of a total of 718 patients randomized in the study, 575 (80%) patients had MMR-proficient (pMMR) tumour status and 143 (20%) patients had MMR-deficient (dMMR) tumour status.
Patients with MMR-deficient (dMMR) endometrial cancer
Among patients with dMMR tumour status, demographic and baseline characteristics were generally well balanced between the treatment arms. Baseline demographics across all three arms were as follows: median age of 62 years (range: 34 to 85), 41% age 65 or older, 1.5% age 75 or older, 62% White, 29% Asian, and 2% Black or African American. Disease characteristics were as follows: ECOG PS of 0 (58%) or 1 (42%), 46% newly diagnosed and 54% recurrent disease. The histologic subtypes were endometrioid (83%), mixed epithelial (5%), serous (3%), carcinosarcoma (3%), undifferentiated (2%), and other (3%).
In patients with dMMR tumour status, the results are summarised in Table 12 and Figure 19. The median follow-up time for PFS in censored patients with dMMR tumour status was 15.5 months in the platinum-based chemotherapy + IMFINZI arm and 10.2 months in the platinum-based chemotherapy arm. At the time of PFS analysis, interim OS data were 26% mature with events in 25 of 95 patients treated with platinum-based chemotherapy + IMFINZI and platinum-based chemotherapy.
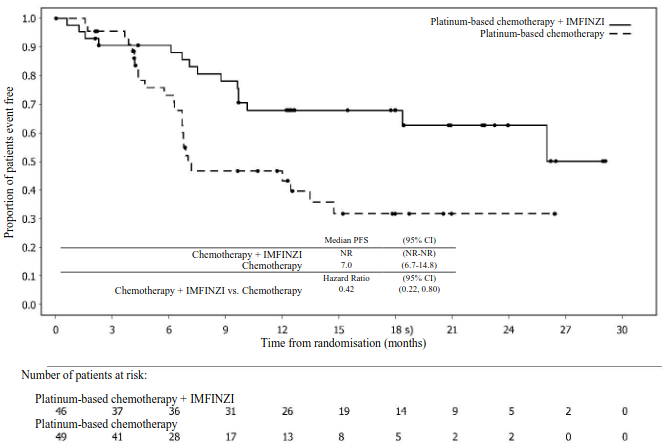
Table 12. Efficacy results for the DUO-E Study (Patients with dMMR tumour status):
| Platinum-based chemotherapy + IMFINZI N=46 | Platinum-based chemotherapy N=49 | |
|---|---|---|
| PFSa,b | ||
| Number of events (%) | 15 (32.6) | 25 (51.0) |
| Median PFS (months) (95% CI)c | NR (NR, NR) | 7.0 (6.7, 14.8) |
| HR (95% CI) | 0.42 (0.22, 0.80) | - |
| OSb | ||
| Number of events (%) | 7 (15.2) | 18 (36.7) |
| Median OS (months) (95% CI)c | NR (NR, NR) | 23.7 (16.9, NR) |
| HR (95% CI) | 0.34 (0.13, 0.79) | - |
| ORRb | ||
| ORRd n (%) | 30 (71.4) | 17 (40.5) |
| DoRb | ||
| Median DoR (months) (95% CI)c | NR (NR, NR) | 10.5 (4.3, NR) |
a Investigator assessed.
b Results are based on the first interim analysis (DCO: 12 April 2023).
c Calculated using the Kaplan-Meier technique.
d Response: Best objective response as confirmed complete response or partial response. Based on number of patients in treatment group with measurable disease at baseline (N=42 in platinum-based chemotherapy + IMFINZI arm, N=42 in platinum-based chemotherapy arm).
CI=Confidence Interval, HR=Hazard Ratio, NR=Not Reached
Figure 19. Kaplan-Meier curve of PFS in DUO-E (Patients with dMMR tumour status):
Patients with MMR-proficient (pMMR) endometrial cancer
Among patients with pMMR tumour status, demographic and baseline characteristics were generally well balanced between the treatment arms. Baseline demographics across all three arms were as follows: median age of 64 years (range: 22 to 86), 48% age 65 or older, 8.1% age 75 or older, 56% White, 30% Asian, and 6% Black or African American. Disease characteristics were as follows: ECOG PS of 0 (69%) or 1 (31%), 47% newly diagnosed and 53% recurrent disease. The histologic subtypes were endometrioid (54%), serous (26%), carcinosarcoma (8%), mixed epithelial (4%), clear cell (3%), undifferentiated (2%), mucinous (<1%), and other (3%).
Results in patients with pMMR tumour status are summarised in Table 13 and Figure 20. The median follow-up time in censored patients with pMMR tumour status was 15.2 months in the platinum-based chemotherapy + IMFINZI + olaparib arm, and 12.8 months in the platinum-based chemotherapy arm.
At the time of PFS analysis, interim OS data were 29% mature with events in 110 of 383 patients treated with platinum-based chemotherapy + IMFINZI + olaparib and platinum-based chemotherapy.
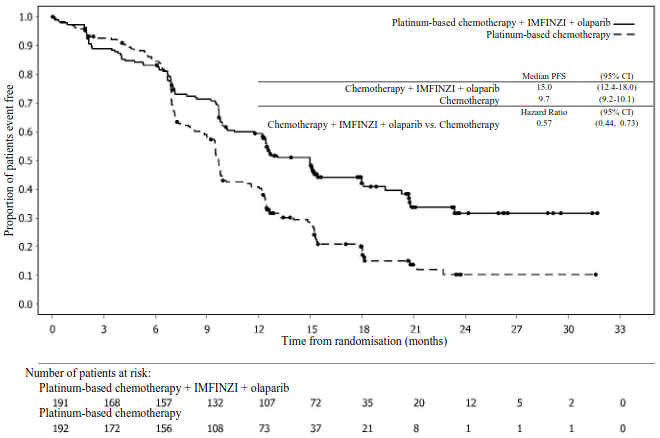
Table 13. Efficacy results for the DUO-E Study (Patients with pMMR tumour status):
| Platinum-based chemotherapy + IMFINZI + olaparib N=191 | Platinum-based chemotherapy N=192 | |
|---|---|---|
| PFSa,b | ||
| Number of events (%) | 108 (56.5) | 148 (77.1) |
| Median PFS (months) (95% CI)c | 15.0 (12.4, 18.0) | 9.7 (9.2, 10.1) |
| HR (95% CI) | 0.57 (0.44, 0.73) | - |
| OSb | ||
| Number of events (%) | 46 (24.1) | 64 (33.3) |
| Median OS (months) (95% CI)c | NR (NR, NR) | 25.9 (25.1, NR) |
| HR (95% CI) | 0.69 (0.47, 1.00) | - |
| ORRb | ||
| ORRd n (%) | 90 (61.2) | 92 (59.0) |
| DoRb | ||
| Median DoR (months) (95% CI)c | 18.7 (10.5, NR) | 7.6 (7.1, 10.2) |
a Investigator assessed.
b Results are based on the first interim analysis (DCO: 12 April 2023).
c Calculated using the Kaplan-Meier technique.
d Response: Best objective response as confirmed complete response or partial response. Based on number of patients in treatment group with measurable disease at baseline (N=147 in platinum-based chemotherapy + IMFINZI + olaparib arm, N=156 in platinum-based chemotherapy arm).
CI=Confidence Interval, HR=Hazard Ratio, NR=Not Reached
Figure 20. Kaplan-Meier curve of PFS in DUO-E (Patients with pMMR tumour status):
Among patients with pMMR tumour status, the PFS HRs were 0.44 (95% CI: 0.31, 0.61) in patients with PD-L1 expression positive status (236/383; 62%) and 0.87 (95% CI: 0.59, 1.28) in patients with PD-L1 expression negative status (140/383; 37%), for the platinum-based chemotherapy + IMFINZI + olaparib arm compared to the platinum-based chemotherapy arm. PD-L1 expression positive was defined as tumour area positive (TAP) ≥1%.
Muscle invasive bladder cancer (MIBC) – NIAGARA Study
NIAGARA was a randomised, open-label, multicentre Phase III study designed to evaluate the efficacy of neoadjuvant IMFINZI in combination with gemcitabine and cisplatin followed by adjuvant IMFINZI monotherapy in patients with MIBC. The study randomised 1 063 patients who were candidates for radical cystectomy and had not received prior systemic chemotherapy or immune-mediated therapy for the treatment of MIBC with clinical tumour stage T2-T4aN0/1M0. The study excluded patients with pure non-urothelial histology, any small cell histology and primary non-bladder (i.e, ureter, urethral, or renal pelvis) cancer of the urothelium, active or prior documented autoimmune disease, active tuberculosis or hepatitis B or C or HIV infection, or use of immuno-suppressive medication within 14 days of the first dose of durvalumab except systemic corticosteroids when used at physiological doses or as premedication.
Randomisation was stratified by clinical tumour stage T2N0 vs. > T2N0 (including T2N1, T3, and T4a), renal function (adequate renal function: creatinine clearance [CrCl] ≥60 mL/min vs. borderline renal function: CrCl ≥40 mL/min to <60 mL/min), and PD-L1 expression (high vs. low/negative) status. Patients were randomised 1:1 to receive perioperative IMFINZI with neoadjuvant chemotherapy (Arm 1) or neoadjuvant chemotherapy alone (Arm 2):
- Arm 1 (IMFINZI + chemotherapy): IMFINZI 1 500 mg + gemcitabine 1 000 mg/m² and cisplatin 70 mg/m² every 3 weeks for 4 cycles prior to surgery, followed by IMFINZI 1 500 mg every 4 weeks for up to 8 cycles after surgery, or
- Arm 2 (Chemotherapy): Gemcitabine 1 000 mg/m² and cisplatin 70 mg/m² every 3 weeks for 4 cycles prior to surgery, without post-surgery treatment.
Patients with borderline renal function received split dose cisplatin of 35 mg/m² on days 1 and 8 of each cycle.
A RECIST 1.1 tumour assessment was performed at baseline and upon completion of neoadjuvant therapy (prior to surgery). After surgery, RECIST 1.1 tumour assessments were performed every 12 weeks for the first 24 months, then every 24 weeks for 36 months, and then every 52 weeks thereafter until progression, the end of study, or death.
The primary endpoints were pathological complete response (pCR) by blinded central pathology review and event-free survival (EFS) which included blinded independent central review (BICR) assessment. Overall survival (OS) was a key secondary endpoint.
The demographics and baseline disease characteristics were generally well-balanced between the 533 patients in Arm 1 and 530 patients in Arm 2. Baseline demographics were as follows: male (81.8%), age <65 years (46.9%), white (67%), Asian (27.9%), black or African American (0.9%), other (0.8%), Hispanic or Latino (8.0%), and ECOG PS 0 (78%) vs. PS 1 (22%). Disease characteristics were as follows: Tumour Stage T2N0 (40.3%) and > T2N0a (59.7%), regional lymph nodes N0 (94.5%) and N1 (5.5%), adequate renal function (81.1%) and borderline renal function (18.9%), and PD-L1 expression status high (73.1%) and low/negative (26.9%). The histologic subtypes included urothelial carcinoma (84.5%), urothelial carcinoma with squamous differentiation (8.2%), urothelial carcinoma with variant histology (5.0%), and urothelial carcinoma with glandular
differentiation (2.4%).
In the overall population, 469 (88.0%) patients in Arm 1 and 441 (83.2%) patients in Arm 2 underwent radical cystectomy.
Results are presented in Table 14, Figure 21 and Figure 22.
Table 14. Efficacy Results for the NIAGARA Study:
| IMFINZI + chemotherapy (N=533) | Chemotherapy (N=530) | |
|---|---|---|
| EFSa | ||
| Number of events (%) | 187 (35.1) | 246 (46.4) |
| Median EFS (months) (95% CI)b | NR (NR, NR) | 46.1 (32.2, NR) |
| HR (95% CI)c | 0.68 (0.56, 0.82) | |
| 2-sided p-valued,e | <0.0001 | |
| pCRf | ||
| Number of patients with response | 180 | 137 |
| Response rate, % (95% CI)g | 33.8 (29.8, 38.0) | 25.8 (22.2, 29.8) |
| Odds ratio (95% CI)h | 1.49 (1.14, 1.96) | |
| 2-sided p-valueh | 0.0038 | |
| OSa | ||
| Number of events (%) | 136 (25.5) | 169 (31.9) |
| Median OS (months) (95% CI)b | NR (NR, NR) | NR (NR, NR) |
| HR (95% CI)c | 0.75 (0.59, 0.93) | |
| 2-sided p-valued,e | 0.0106 | |
a Results are based on a pre-planned interim analysis (DCO: 29 April 2024) which occurred 68 months after study initiation.
b Calculated using the Kaplan-Meier technique.
c Based on stratified Cox proportional hazard model with tumour stage [T2N0 vs. > T2N0], renal function [adequate vs. borderline], and PD-L1 status [high vs. low/negative] as stratification factors.
d Based on stratified log-rank test with tumour stage [T2N0 vs. > T2N0], renal function [adequate vs borderline], and PD-L1 status [high vs low/negative] as stratification factors.
e The boundary for declaring statistical significance for the primary efficacy endpoints pCR rate, EFS and the key secondary endpoint OS were determined by a multiple test procedure with an alpha-exhaustive recycling strategy. Alpha allocated to EFS and OS at the interim analysis was based on a Lan-DeMets alpha spending function with O'Brien Fleming approach (pCR = 0.001, EFS = 0.0412, OS = 0.0154, 2-sided).
f Based on the final analysis of pCR (DCO: 14 Jan 2022).
g CI was calculated using the Clopper Pearson method.
h Obtained using logistic regression adjusted for the stratification factors (renal function [adequate vs. borderline], tumour stage [T2N0 vs. > T2N0] and PD-L1 status [high vs. low/negative] per IVRS).
CI=Confidence Interval, HR=Hazard Ratio, NR=Not Reached
Figure 21. Kaplan-Meier Curve of EFS:


Subgroup analysis
In an exploratory analysis by tumour stage, the EFS HR was 0.61 (95% CI: 0.48, 0.78) in the subgroup of patients with clinical stage > T2N0 (N=635) and 0.81 (95% CI: 0.60, 1.10) in the subgroup of patients with clinical stage T2N0 (N=428). The OS HR was 0.67 (95% CI: 0.50, 0.89) in the subgroup of patients with clinical stage > T2N0 and 0.89 (95% CI: 0.62, 1.29) in patients with clinical stage T2N0.
Resectable GC/GEJC – MATTERHORN Study
MATTERHORN was a randomised, double-blind, placebo-controlled, multicentre, Phase III study to evaluate the efficacy of IMFINZI in combination with FLOT (5-FU, leucovorin, oxaliplatin, docetaxel) chemotherapy as neoadjuvant and adjuvant treatment, followed by adjuvant IMFINZI monotherapy, in patients with resectable gastric or gastro-oesophageal junction adenocarcinoma (Stage IIA to Stage IVA [AJCC, 8th edition]). The study enrolled previously untreated patients with documented GC/GEJC, with no prior exposure to immune-mediated therapy, and WHO/ECOG performance status of 0 or 1. Patients were enrolled regardless of PD-L1 expression level. Prior to randomisation, patients had tumour PD-L1 expression level confirmed using the TAP scoring method, which is defined as the total percentage of the tumour area (tumour and any desmoplastic stroma) covered by tumour cells with PD-L1 membrane staining at any intensity and tumour-associated immune cells with PD-L1 staining at any intensity as visually estimated, by the Ventana PD-L1 (SP263) Assay. The study excluded patients with active or prior documented autoimmune or inflammatory disorders, or use of immunosuppressive medication within 14 days of the first dose of durvalumab.
Randomisation was stratified by geographic region (Asia vs. non-Asia), clinical lymph node status (positive vs. negative), and PD-L1 expression level (TAP <1% vs. TAP ≥1%).
Patients were randomised in a 1:1 ratio to one of the following treatment arms. Crossover between the study arms was not permitted.
- Arm 1: IMFINZI 1 500 mg on Day 1 + FLOT (5-FU 2 600 mg/m², Leucovorin 200 mg/m², Oxaliplatin 85 mg/m², Docetaxel 50 mg/m²) chemotherapy on Days 1 and 15 every 4 weeks for 4 cycles (1 dose of IMFINZI and 2 doses of FLOT per cycle; 2 cycles in the neoadjuvant phase + 2 cycles in the adjuvant phase) followed by IMFINZI 1 500 mg on Day 1 every 4 weeks for up to 10 additional cycles post-surgery for a total of 12 cycles (1 dose per cycle)
- Arm 2: Placebo on Day 1 + FLOT (5-FU 2 600 mg/m², Leucovorin 200 mg/m², Oxaliplatin 85 mg/m², Docetaxel 50 mg/m²) chemotherapy on Days 1 and 15 every 4 weeks for 4 cycles (1 dose of placebo and 2 doses of FLOT per cycle; 2 cycles in the neoadjuvant phase + 2 cycles in the adjuvant phase) followed by placebo on Day 1 every 4 weeks for up to 10 additional cycles post-surgery for a total of 12 cycles (1 dose per cycle)
Patients in either the neoadjuvant or adjuvant phase who discontinued FLOT chemotherapy due to reasons other than disease progression or recurrence could, based on investigator discretion, continue treatment with IMFINZI monotherapy as described above.
A baseline RECIST 1.1 tumour assessment was performed prior to the start of neoadjuvant therapy, with a follow-up scan prior to surgery within 4 weeks after the final chemotherapy dose. An adjuvant baseline scan was performed no sooner than 4 weeks after surgery and before starting the adjuvant treatment. Tumour assessments were conducted every 12 weeks (relative to the adjuvant baseline scan) for 2 years, then every 24 weeks until RECIST 1.1 defined radiological progression, consent withdrawal, or death.
The primary endpoint of the study was EFS by BICR assessment, defined as the time from randomisation until the date of one of the following events (whichever occurred first): disease progression according to RECIST, version 1.1, as assessed by blinded independent central review (BICR), that precluded surgery or that required non-protocol therapy during the neoadjuvant treatment period; progression or recurrence according to RECIST, version 1.1, during the adjuvant treatment period; non-RECIST progression (according to investigator assessment or confirmed by biopsy) that precluded surgery or that required non-protocol therapy during the neoadjuvant treatment period or that was discovered during surgery; progression or recurrence confirmed by biopsy after surgery; or death from any cause. The key secondary endpoints were OS and pCR rate by blinded central pathology review.
The demographics and baseline disease characteristics were generally well balanced between the two study arms (474 patients in Arm 1 and 474 patients in Arm 2). The baseline demographics of the population were as follows: male (71.9%), age ≥65 years (41.4%), median age 62 years (range: 26 to 84), White (67.8%), Asian (20.4%), Black or African American (1.1%), American Indian or Alaska Native (4.0%), Other Race (1.7%), Not Hispanic or Latino (80.0%), WHO/ECOG PS 0 (74.2%) versus PS 1 (25.8%). Disease characteristics were as follows: Stage II (29.4%), Stage III (61.7%), Stage IVA (8.8%), Gastric (67.5%), Gastro-oesophageal junction (32.5%), Siewert type 1 (10.4%), Siewert type 2 (14.8%), Siewert type 3 (7.3%), Intestinal type (50.9%), Diffuse type (26.3%), Indeterminate type (22.8%), Clinical lymph node status Positive (70.4%), Clinical lymph node status Negative (29.2%), PD-L1 expression status TAP ≥ 1% (90.0%), PD-L1 expression status TAP <1% (10.0%).
There were 431 (90.9%) patients in Arm 1 who attempted curative intent surgery compared to 428 (90.3%) patients in Arm 2. There were 412 (86.9%) patients in Arm 1 who completed curative intent surgery compared to 400 (84.4%) patients in Arm 2.
Table 15. Efficacy Results for the MATTERHORN Study:
| IMFINZI + FLOT chemotherapy (N=474) | Placebo + FLOT chemotherapy (N=474) | |
| EFSa | ||
| Number of events, n (%) | 167 (35.2%) | 218 (46%) |
| Median EFS (months) (95% CI)b | NR (40.7, NR) | 32.8 (27.9, NR) |
| HR (95% CI)c | 0.71 (0.58, 0.86) | |
| 2-sided p-valued,e | <0.001 | |
| EFS at 24 months, % (95% CI)b | 67.4 (62.9, 71.6) | 58.5 (53.8, 63.0) |
| OSf | ||
| Number of deaths (%) | 160 (33.8%) | 192 (40.5%) |
| Median OS (95% CI) (months)b | NR (NR, NR) | NR (NR, NR) |
| HR (95% CI)c | 0.78 (0.63, 0.96) | |
| 2-sided p-valued,g | 0.021 | |
| OS at 24 months, % (95% CI)b | 75.5 (71.4, 79.1) | 70.4 (66.0, 74.3) |
| OS at 36 months, % (95% CI)b | 68.6 (64.2, 72.6) | 61.9 (57.3, 66.2) |
a Results are based on an EFS pre-specified interim analysis (DCO: 20 December 2024).
b Calculated using the Kaplan-Meier technique.
c Based on stratified Cox proportional hazards model stratified by geographical region, clinical lymph node status and PD-L1 expression status at randomisation. An HR <1 favours IMFINZI. CI calculated using the profile likelihood approach.
d Based on stratified log-rank test adjusting for geographic region, clinical lymph node status, and PD-L1 expression status at randomisation.
e Based on a Lan-DeMets alpha spending function with O'Brien Fleming boundary calculated using the actual number of events at the DCO, the boundary for declaring statistical significance of EFS was 0.0239 for an overall alpha of 5% (2-sided).
f Results are based on an OS pre-specified final analysis (DCO: 01 September 2025).
g An alpha of 4.99% (2-sided) was allocated to the final analysis of OS, corresponding to a boundary for declaring statistical significance of 0.0499.
CI=Confidence Interval, HR=Hazard Ratio, NR=Not reached
Figure 23. Kaplan-Meier Curve of EFS:

Figure 24. Kaplan-Meier Curve of OS:

Subgroup analysis
In a predefined exploratory analysis by clinical lymph node status, the EFS HR was 0.67 (95% CI: 0.53, 0.84) in the clinical lymph node positive subgroup (N=667) and 0.85 (95% CI: 0.57, 1.27) in the clinical lymph node negative subgroup (N=277). The OS HR was 0.73 (95% CI: 0.57, 0.94) in the clinical lymph node positive subgroup and 1.01 (95% CI: 0.67, 1.51) in the clinical lymph node negative subgroup.
Paediatric population
The safety and efficacy of IMFINZI in combination with tremelimumab in children and adolescents aged less than 18 years has not been established. Study D419EC00001 was a multi-centre, open-label dose finding and dose expansion study to evaluate the safety, preliminary efficacy and pharmacokinetics of IMFINZI in combination with tremelimumab followed by IMFINZI monotherapy, in paediatric patients with advanced malignant solid tumours (except primary central nervous system tumours) who had disease progression and for whom no standard of care treatment exists. The study enrolled 50 paediatric patients with an age range from 1 to 17 years with primary tumour categories: neuroblastoma, solid tumour and sarcoma. Patients received either IMFINZI 20 mg/kg in combination with tremelimumab 1 mg/kg or IMFINZI 30 mg/kg in combination with tremelimumab 1 mg/kg intravenously every 4 weeks for 4 cycles, followed by IMFINZI as monotherapy every 4 weeks. In the dose finding phase, IMFINZI and tremelimumab combination therapy was preceded by a single cycle of IMFINZI monotherapy; 8 patients in this phase however discontinued treatment prior to receiving tremelimumab. Thus, of the 50 patients enrolled in the study, 42 received IMFINZI in combination with tremelimumab and 8 received IMFINZI only. In the dose-expansion phase, an ORR of 5.0% (1/20 patients) was reported in the evaluable for response analysis set. No new safety signals were observed relative to the known safety profiles of IMFINZI and tremelimumab in adults. See section 4.2 for information on paediatric use.
Pharmacokinetic properties
The pharmacokinetics (PK) of durvalumab was assessed for IMFINZI as a single agent, in combination with chemotherapy, in combination with tremelimumab and platinum-based chemotherapy, in combination with tremelimumab and in combination with platinum-based chemotherapy followed by IMFINZI in combination with olaparib.
The PK of durvalumab was studied in 2903 patients with solid tumours with doses ranging from 0.1 to 20 mg/kg administered intravenously once every two, three or four weeks as monotherapy. PK exposure increased more than dose-proportionally (non-linear PK) at doses <3 mg/kg, and dose proportionally (linear PK) at doses ≥3 mg/kg. Steady state was achieved at approximately 16 weeks. Based on population PK analysis that included 1878 patients who received durvalumab monotherapy in the dose range of ≥10 mg/kg every 2 weeks, the geometric mean steady state volume of distribution (Vss) was 5.64 L. Durvalumab clearance (CL) decreased over time resulting in a geometric mean steady state clearance (CLss) of 8.16 ml/h at Day 365; the decrease in CLss was not considered clinically relevant. The terminal half-life (t1/2), based on baseline CL, was approximately 18 days. There was no clinically meaningful difference between the PK of durvalumab as a single agent, in combination with chemotherapy, in combination with tremelimumab and platinum-based chemotherapy, in combination with tremelimumab and in combination with platinum-based chemotherapy followed by IMFINZI in combination with olaparib. The primary elimination pathways of durvalumab are protein catabolism via reticuloendothelial system or target mediated disposition.
Special populations
Age (19-96 years), body weight (31-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, soluble PD-L1, tumour type, race or ECOG status had no clinically significant effect on the PK of durvalumab.
Renal impairment
Mild (creatinine clearance (CrCL) 60 to 89 ml/min) and moderate renal impairment (creatinine clearance (CrCL) 30 to 59 ml/min) had no clinically significant effect on the PK of durvalumab. The effect of severe renal impairment (CrCL 15 to 29 ml/min) on the PK of durvalumab is unknown; however, as IgG monoclonal antibodies are not primarily cleared via renal pathways, a change in renal function is not expected to influence durvalumab exposure.
Hepatic impairment
Mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin >1.0 to 1.5 x ULN and any AST) or moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST) had no clinically significant effect on the PK of durvalumab. The effect of severe hepatic impairment (bilirubin >3.0 x ULN and any AST) on the pharmacokinetics of durvalumab is unknown; however, as IgG monoclonal antibodies are not primarily cleared via hepatic pathways, a change in hepatic function is not expected to influence durvalumab exposure.
Paediatric population
The PK of durvalumab in combination with tremelimumab was evaluated in a study of 50 paediatric patients with an age range from 1 to 17 years in study D419EC00001. Patients received either durvalumab 20 mg/kg in combination with tremelimumab 1 mg/kg or durvalumab 30 mg/kg in combination with tremelimumab 1 mg/kg intravenously every 4 weeks for 4 cycles, followed by durvalumab as monotherapy every 4 weeks. Based on population PK analysis, durvalumab systemic exposure in paediatric patients ≥35 kg receiving durvalumab 20 mg/kg every 4 weeks was similar to exposure in adults receiving durvalumab 20 mg/kg every 4 weeks, whereas in paediatric patients (≥35 kg) receiving durvalumab 30mg/kg every 4 weeks, exposure was approximately 1.5-fold higher compared to exposure in adults receiving durvalumab 20 mg/kg every 4 weeks. In paediatric patients <35 kg receiving durvalumab 30 mg/kg every 4 weeks, the systemic exposure was similar to exposure in adults receiving durvalumab 20 mg/kg every 4 weeks.
Preclinical safety data
Carcinogenicity and mutagenicity
The carcinogenic and genotoxic potential of durvalumab has not been evaluated.
Reproductive toxicology
As reported in the literature, the PD-1/PD-L1 pathway plays a central role in preserving pregnancy by maintaining maternal immune tolerance to the foetus, and in mouse allogeneic pregnancy models disruption of PD-L1 signalling was shown to result in an increase in foetal loss. In animal reproduction studies, administration of durvalumab to pregnant cynomolgus monkeys from the confirmation of pregnancy through delivery, at exposure levels approximately 18-times higher than those observed at the clinical dose of 10 mg/kg of durvalumab (based on AUC), was associated with placental transfer but not with maternal toxicity or effects on embryofoetal development, pregnancy outcome or postnatal development. Negligible levels of durvalumab was found in milk of cynomolgous monkey on Day 28 after birth.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.