IMJUDO Concentrate for solution for infusion Ref.[50776] Active ingredients: Tremelimumab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other monoclonal antibodies and antibody drug conjugates
ATC code: L01FX20
Mechanism of action
Cytotoxic T lymphocyte-associated antigen (CTLA-4) is primarily expressed on the surface of T lymphocytes. Interaction of CTLA-4 with its ligands, CD80 and CD86, limits effector T-cell activation, through a number of potential mechanisms, but primarily by limiting co-stimulatory signalling through CD28.
Tremelimumab is a selective, fully human IgG2 antibody that blocks CTLA-4 interaction with CD80 and CD86, thus enhancing T-cell activation and proliferation, resulting in increased T-cell diversity and enhanced anti-tumour activity.
The combination of tremelimumab, a CTLA-4 inhibitor and durvalumab, a PD-L1 inhibitor results in improved anti-tumour responses in metastatic non-small cell lung cancer and hepatocellular carcinoma.
Clinical efficacy
HCC - HIMALAYA Study
The efficacy of IMJUDO 300 mg as a single dose in combination with durvalumab was evaluated in the HIMALAYA Study, a randomised, open-label, multicentre study in patients with confirmed uHCC who did not receive prior systemic treatment for HCC. The study included patients with Barcelona Clinic Liver Cancer (BCLC) Stage C or B (not eligible for locoregional therapy) and Child-Pugh Score Class A.
The study excluded patients with brain metastases or a history of brain metastases, co-infection of viral hepatitis B and hepatitis C; active or prior documented gastro-intestinal (GI) bleeding within 12 months; ascites requiring non-pharmacologic intervention within 6 months; hepatic encephalopathy within 12 months before the start of treatment; active or prior documented autoimmune or inflammatory disorders.
Patients with esophageal varices were included except those with active or prior documented GI bleeding within 12 months prior to study entry.
Randomisation was stratified by macrovascular invasion (MVI) (yes vs. no), aetiology of liver disease (confirmed hepatitis B virus vs. confirmed hepatitis C virus vs. others) and ECOG performance status (0 vs. 1). The HIMALAYA study randomized 1171 patients 1:1:1 to receive:
- Durvalumab 1500 mg every 4 weeks
- IMJUDO 300 mg as a single dose + durvalumab 1500 mg; followed by durvalumab 1500 mg every 4 weeks
- Sorafenib 400 mg twice daily
The primary endpoint was Overall Survival (OS) for the comparison of IMJUDO 300 mg as a single dose in combination with durvalumab vs. sorafenib. Secondary endpoints included Progression-Free Survival (PFS), Investigator-assessed Objective Response Rate (ORR) and Duration of Response (DoR) according to RECIST v1.1.
The demographics and baseline disease characteristics were well balanced between study arms. The baseline demographics of the overall study population were as follows: male (83.7%), age < 65 years (50.4%), White (44.6%), Asian (50.7%), Black or African American (1.7%), Other race (2.3%), ECOG PS 0 (62.6%); Child-Pugh Class score A (99.5%), macrovascular invasion (25.2%), extrahepatic spread (53.4%), baseline AFP < 400 ng/ml (63.7%), baseline AFP ≥400 ng/ml (34.5%), viral aetiology; hepatitis B (30.6%), hepatitis C (27.2%), uninfected (42.2%), evaluable PD-L1 data (86.3%), PD-L1 Tumour area positivity (TAP) ≥1% (38.9%), PD-L1 TAP < 1% (48.3%) [Ventana PD-L1 (SP263) assay].
Results are presented in Table 4 and Figure 1.
Table 4. Efficacy results for the HIMALAYA study for IMJUDO 300 mg with durvalumab vs. Sorafenib:
| IMJUDO 300 mg + durvalumab (n=393) | S (n=389) | |
|---|---|---|
| Follow-up duration | ||
| Median follow-up (months)a | 33.2 | 32.2 |
| OS | ||
| Number of deaths (%) | 262 (66.7) | 293 (75.3) |
| Median OS (months) (95% CI) | 16.4 (14.2, 19.6) | 13.8 (12.3, 16.1) |
| HR (95% CI) | 0.78 (0.66, 0.92) | |
| p-valueb | 0.0035 | |
| PFS | ||
| Number of events (%) | 335 (85.2) | 327 (84.1) |
| Median PFS (months) (95% CI) | 3.78 (3.68, 5.32) | 4.07 (3.75, 5.49) |
| HR (95% CI) | 0.90 (0.77, 1.05) | |
| ORR | ||
| ORR n (%)c | 79 (20.1) | 20 (5.1) |
| Complete Response n (%) | 12 (3.1) | 0 |
| Partial Response n (%) | 67 (17.0) | 20 (5.1) |
| DoR | ||
| Median DoR (months) | 22.3 | 18.4 |
a Calculated using the reverse Kaplan-Meier technique (with censor indicator reversed).
b Based on a Lan-DeMets alpha spending function with O'Brien Fleming type boundary and the actual number of events observed, the boundary for declaring statistical significance for IMJUDO 300 mg + durvalumab vs. Sorafenib was 0.0398 (Lan◦and◦DeMets 1983).
c Confirmed complete response.
CI=Confidence Interval
Figure 1. Kaplan-Meier curve of OS:

NSCLC – POSEIDON study
POSEIDON was a study designed to evaluate the efficacy of durvalumab with or without IMJUDO in combination with platinum-based chemotherapy. POSEIDON was a randomised, open-label, multicentre study in 1013 metastatic NSCLC patients with no sensitising epidermal growth factor receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) genomic tumour aberrations. Patients with histologically or cytologically documented metastatic NSCLC were eligible for enrolment. Patients had no prior chemotherapy or any other systemic therapy for metastatic NSCLC. Prior to randomisation, patients had tumour PD-L1 status confirmed by using the Ventana PD-L1 (SP263) assay. Patients had a World Health Organization (WHO)/Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 at enrolment.
The study excluded patients with active or prior documented autoimmune disease; active and/or untreated brain metastases; a history of immunodeficiency; administration of systemic immunosuppression within 14 days before the start of IMJUDO or durvalumab, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection; or patients receiving live attenuated vaccine within 30 days before or after the start of IMJUDO and/or durvalumab (see section 4.4).
Randomisation was stratified by tumour cells (TC) PD-L1 expression (TC ≥50% vs. TC <50%), disease stage (Stage IVA vs. Stage IVB, per the 8th edition of American Joint Committee on Cancer), and histology (non-squamous vs. squamous).
Patients were randomised 1:1:1 to receive:
- Arm 1: IMJUDO 75 mg with durvalumab 1500 mg and platinum-based chemotherapy every 3 weeks for 4 cycles, followed by durvalumab 1500 mg every 4 weeks as monotherapy. A fifth dose of IMJUDO 75 mg was given at Week 16 alongside durvalumab dose 6.
- Arm 2: Durvalumab 1500 mg and platinum-based chemotherapy every 3 weeks for 4 cycles, followed by durvalumab 1500 mg every 4 weeks as monotherapy.
- Arm 3: Platinum-based chemotherapy every 3 weeks for 4 cycles. Patients could receive 2 additional cycles (a total of 6 cycles post-randomisation), as clinically indicated, at investigator's discretion.
Patients received one of the following platinum-based chemotherapy regimens:
- Non-squamous NSCLC
- Pemetrexed 500 mg/m² with carboplatin AUC 5-6 or cisplatin 75 mg/m² every 3 weeks. Unless contraindicated by the investigator, pemetrexed maintenance could be given.
- Squamous NSCLC
- Gemcitabine 1000 or 1250 mg/m² on Days 1 and 8 with cisplatin 75 mg/m² or carboplatin AUC 5-6 on Day 1 every 3 weeks.
- Non-squamous or squamous NSCLC
- Nab-paclitaxel 100 mg/m² on Days 1, 8, and 15 with carboplatin AUC 5-6 on Day 1 every 3 weeks.
IMJUDO was given up to a maximum of 5 doses unless there was disease progression or unacceptable toxicity. Durvalumab and histology-based pemetrexed maintenance therapy (when applicable) was continued until disease progression or unacceptable toxicity.
Tumour assessments were conducted at Week 6 and Week 12 from the date of randomisation, and then every 8 weeks until confirmed objective disease progression. Survival assessments were conducted every 2 months following treatment discontinuation.
The dual primary endpoints of the study were progression-free survival (PFS) and overall survival (OS) for durvalumab + platinum-based chemotherapy (Arm 2) vs. platinum-based chemotherapy alone (Arm 3). The key secondary endpoints of the study were PFS and OS for IMJUDO + durvalumab + platinum-based chemotherapy (Arm 1) and platinum-based chemotherapy alone (Arm 3). The secondary endpoints included objective response rate (ORR) and duration of response (DoR). PFS, ORR, and DoR were assessed using Blinded Independent Central Review (BICR) according to RECIST v1.1.
The demographics and baseline disease characteristics were well-balanced between study arms. Baseline demographics of the overall study population were as follows: male (76.0%), age ≥65 years (47.1%), age ≥75 years (11.3%) median age 64 years (range: 27 to 87 years), White (55.9%), Asian (34.6%), Black or African American (2.0%), other (7.6%), non-Hispanic or Latino (84.2%), current smoker or past-smoker (78.0%), WHO/ECOG PS 0 (33.4%) and WHO/ECOG PS 1 (66.5%). Disease characteristics were as follows: Stage IVA (50.0%), Stage IVB (49.6%), histological sub-groups of squamous (36.9%), non-squamous (62.9%), brain metastases (10.5%), PD-L1 expression TC ≥50% (28.8%) and PD-L1 expression TC <50% (71.1%).
The study showed a statistically significant improvement in OS with IMJUDO + durvalumab + platinum-based chemotherapy (Arm 1) vs. platinum-based chemotherapy alone (Arm 3). IMJUDO + durvalumab + platinum-based chemotherapy showed a statistically significant improvement in PFS vs. platinum-based chemotherapy alone. The results are summarised below.
Table 5. Efficacy results for the POSEIDON study:
| Arm 1: IMJUDO+durvalumab+ platinum-based chemotherapy (n=338) | Arm 3: Platinum-based chemotherapy (n=337) | |
|---|---|---|
| OSa | ||
| Number of deaths (%) | 251 (74.3) | 285 (84.6) |
| Median OS (months) (95% CI) | 14.0 (11.7, 16.1) | 11.7 (10.5, 13.1) |
| HR (95% CI)b | 0.77 (0.650, 0.916) | |
| p-valuec | 0.00304 | |
| PFSa | ||
| Number of events (%) | 238 (70.4) | 258 (76.6) |
| Median PFS (months) (95% CI) | 6.2 (5.0, 6.5) | 4.8 (4.6, 5.8) |
| HR (95% CI)b | 0.72 (0.600, 0.860) | |
| p-valuec | 0.00031 | |
| ORR n (%)d,e | 130 (38.8) | 81 (24.4) |
| Complete Response n (%) | 2 (0.6) | 0 |
| Partial Response n (%) | 128 (38.2) | 81 (24.4) |
| Median DoR (months) (95% CI)d,e | 9.5 (7.2, NR) | 5.1 (4.4, 6.0) |
a Analysis of PFS at data cut off 24 July 2019 (median follow up 10.15 months). Analysis of OS at data cut off 12 March 2021 (median follow up 34.86 months). The boundaries for declaring efficacy (Arm 1 vs. Arm 3: PFS 0.00735, OS 0.00797; 2-sided) were determined by a Lan-DeMets alpha spending function that approximates an O'Brien Fleming approach. PFS was assessed by BICR according to RECIST v1.1.
b HR are derived using a Cox pH model stratified by PD-L1, histology and disease stage.
c 2-sided p-value based on a log-rank test stratified by PD-L1, histology and disease stage.
d Confirmed Objective Response.
e Post-hoc analysis.
NR=Not Reached, CI=Confidence Interval
Figure 2. Kaplan-Meier curve of OS:
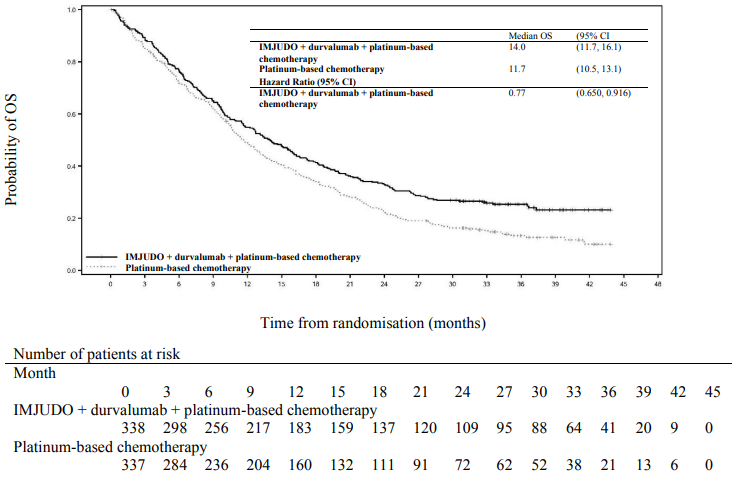
Figure 3. Kaplan-Meier curve of PFS:
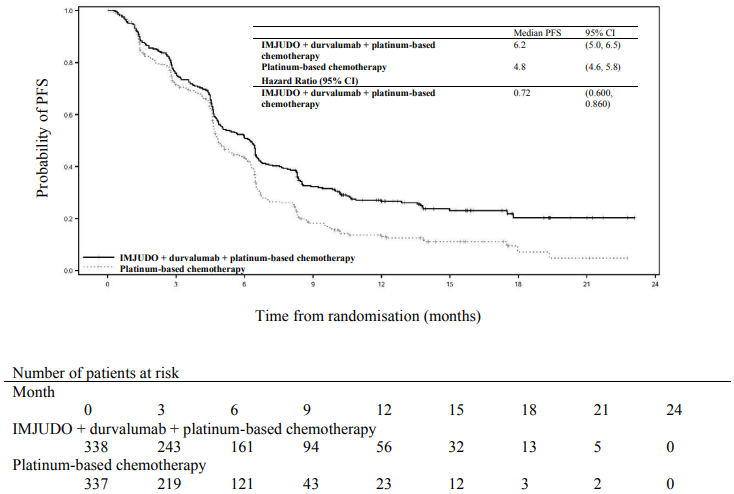
Figure 4 summarises efficacy results of OS by tumour PD-L1 expression in prespecified subgroup analyses.
Figure 4. Forest plot of OS by PD-L1 expression for IMJUDO + durvalumab + platinum-based chemotherapy vs. platinum-based chemotherapy:
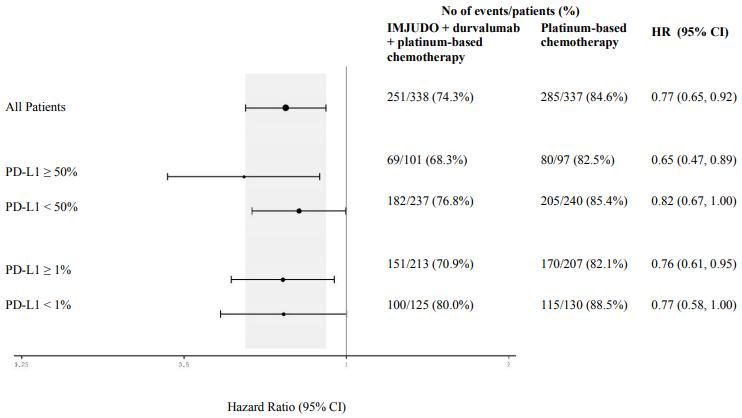
Elderly population
A total of 75 patients aged ≥75 years were enrolled in the IMJUDO in combination with durvalumab and platinum-based chemotherapy (n=35) and platinum-based chemotherapy only (n=40) arms of the POSEIDON study. An exploratory HR of 1.05 (95% CI: 0.64, 1.71) for OS was observed for IMJUDO in combination with durvalumab and platinum-based chemotherapy vs. platinum-based chemotherapy within this study subgroup. Due to the exploratory nature of this subgroup analysis no definitive conclusions can be drawn, but caution is suggested when considering this regimen for elderly patients.
Paediatric population
The safety and efficacy of IMJUDO in combination with durvalumab in children and adolescents aged less than 18 years has not been established. Study D419EC00001 was a multi centre, open-label dose finding and dose expansion study to evaluate the safety, preliminary efficacy and pharmacokinetics of IMJUDO in combination with durvalumab followed by durvalumab monotherapy in paediatric patients with advanced malignant solid tumours (except primary central nervous system tumours) who had disease progression and for whom no standard of care treatment exists. The study enrolled 50 paediatric patients with an age range from 1 to 17 years with primary tumour categories: neuroblastoma, solid tumour and sarcoma. Patients received IMJUDO 1 mg/kg either in combination with durvalumab 20 mg/kg or durvalumab 30 mg/kg every 4 weeks for 4 cycles, followed by durvalumab as monotherapy every 4 weeks. In the dose finding phase, IMJUDO and durvalumab combination therapy was preceded by a single cycle of durvalumab; 8 patients in this phase however discontinued treatment prior to receiving IMJUDO. Thus, of the 50 patients enrolled in the study, 42 received IMJUDO in combination with durvalumab and 8 received durvalumab only. In the doseexpansion phase, an ORR of 5.0% (1/20 patients) was reported in the evaluable for response analysis set. No new safety signals were observed relative to the known safety profiles of IMJUDO and durvalumab in adults. See section 4.2 for information on paediatric use.
5.2. Pharmacokinetic properties
The pharmacokinetics (PK) of tremelimumab was assessed for tremelimumab as monotherapy,in combination with durvalumab and in combination with platinum-based chemotherapy.
The PK of tremelimumab was studied in patients with doses ranging from 75 mg to 750 mg or 10 mg/kg administered intravenously once every 4 or 12 weeks as monotherapy, or at a single dose of 300 mg. PK exposure increased dose proportionally (linear PK) at doses ≥75 mg. Steady state was achieved at approximately 12 weeks. Based on population PK analysis that included patients (n=1605) who received tremelimumab monotherapy or in combination with other medicinal products in the dose range of ≥75 mg (or 1 mg/kg) every 3 or 4 weeks, the estimated tremelimumab clearance (CL) and volume of distribution (Vd) were 0.309 l/day and 6.33 l, respectively. The terminal half-life was approximately 14.2 days. The primary elimination pathways of tremelimumab are protein catabolism via reticuloendothelial system or target mediated disposition.
Special populations
Age (18–87 years), body weight (34-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, tumour type, race or ECOG/WHO status had no clinically significant effect on the PK of tremelimumab.
Renal impairment
Mild (creatinine clearance (CrCL) 60 to 89 ml/min) and moderate renal impairment (creatinine clearance (CrCL) 30 to 59 ml/min) had no clinically significant effect on the PK of tremelimumab. The effect of severe renal impairment (CrCL 15 to 29 ml/min) on the PK of tremelimumab is unknown; the potential need for dose adjustment cannot be determined. However, as IgG monoclonal antibodies are not primarily cleared via renal pathways, a change in renal function is not expected to influence tremelimumab exposure.
Hepatic impairment
Mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin >1.0 to 1.5 × ULN and any AST) and moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST) had no clinically significant effect on the PK of tremelimumab. The effect of severe hepatic impairment (bilirubin >3.0 x ULN and any AST) on the PK of tremelimumab is unknown; the potential need for dose adjustment cannot be determined. However, as IgG monoclonal antibodies are not primarily cleared via hepatic pathways, a change in hepatic function is not expected to influence tremelimumab exposure.
Paediatric population
The PK of tremelimumab in combination with durvalumab was evaluated in a study of 50 paediatric patients with an age range from 1 to 17 years in study D419EC00001. Patients received tremelimumab 1 mg/kg either in combination with durvalumab 20 mg/kg or in combination with durvalumab 30 mg/kg every 4 weeks for 4 cycles, followed by durvalumab as monotherapy every 4 weeks. Based on population PK analysis, tremelimumab systemic exposure in paediatric patients ≥35 kg receiving tremelimumab 1 mg/kg every 4 weeks was similar to exposure in adults receiving 1 mg/kg every 4 weeks, whereas in paediatric patients <35 kg, exposure was lower relative to adults.
5.3. Preclinical safety data
Animal toxicology
In the chronic 6-month study in cynomolgus monkeys, treatment with tremelimumab was associated with dose-related incidence in persistent diarrhoea and skin rash, scabs and open sores, which were dose-limiting. These clinical signs were also associated with decreased appetite and body weight and swollen peripheral lymph nodes. Histopathological findings correlating with the observed clinical signs included reversible chronic inflammation in the cecum and colon, mononuclear cell infiltration in the skin and hyperplasia in lymphoid tissues.
A dose-dependent increase in the incidence and severity of mononuclear cell infiltration with or without mononuclear cell inflammation was observed in the salivary gland, pancreas (acinar), thyroid, parathyroid, adrenal, heart, esophagus, tongue, periportal liver area, skeletal muscle, prostate, uterus, pituitary, eye (conjunctiva, extra ocular muscles), and choroid plexus of the brain. No NOAEL was found in this study with animals treated with the lowest dose of 5 mg/kg/week, however the intermediate dose of 15 mg/kg week was considered the highest non-severely toxic dose (HNSTD). This dose provided an exposure-based safety margin of 1.77-5.33 to clinical relevant exposure based on the clinical dosing regimen of either a 300 mg single dose or 75 mg every three weeks.
Carcinogenicity and mutagenicity
The carcinogenic and genotoxic potential of tremelimumab has not been evaluated.
Reproductive toxicology
Mononuclear cell infiltration in prostate and uterus was observed in repeat dose toxicity studies. Since animal fertility studies have not been conducted with tremelimumab, the relevance of these findings for fertility is unknown. In reproduction studies, administration of tremelimumab to pregnant cynomolgus monkeys during the period of organogenesis was not associated with maternal toxicity or effects on pregnancy losses, foetal weights, or external, visceral, skeletal abnormalities or weights of selected foetal organs.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.