IQIRVO Film-coated tablet Ref.[113546] Active ingredients: Elafibranor
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Ipsen Pharma, 65 quai George Gorse, 92100 Boulogne-Billancourt, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Bile and liver therapy, Other drugs for bile therapy
ATC code: A05AX06
Mechanism of action
Elafibranor and its main active metabolite GFT1007 are dual peroxisome proliferator-activated receptor (PPAR)α/δ agonists. PPAR α/δ are thought to be key regulators of bile acid (BA) homeostasis, inflammation and fibrosis.
Activation of PPARα and PPARδ decreases bile toxicity and improve cholestasis by modulating BA synthesis, detoxification and transporters. Activation of PPARα and PPARδ also has anti-inflammatory effects by acting on different pathways.
Pharmacodynamic effects
In the pivotal phase 3 ELATIVE study, treatment with elafibranor resulted in a marked reduction from baseline in alkaline phosphatase (ALP) as early as 4 weeks which was sustained through week 52. In alignment with the observed biochemical response, greater reductions in biomarkers of BA synthesis including the BA precursor 7 alpha-hydroxy-4-cholesten-3-one (C4) and Fibroblast Growth Factor-19 (FGF-19), a BA synthesis regulator, were observed with elafibranor treatment.
Cardiac electrophysiology
Thorough QT (TQT) analysis excluded any prolongation effect of elafibranor on QT/corrected QT (QTc) interval at repeat doses of up to 300 mg for 14 days. In clinical studies, no clinically meaningful changes in vital signs or in electrocardiogram (ECG) (including QTc interval) were observed in participants treated with elafibranor.
Clinical efficacy
The efficacy of elafibranor was evaluated in Study GFT505B-319-1 (ELATIVE), a phase 3, randomised, double blind (DB), placebo-controlled study in 161 adults with PBC with an inadequate response or intolerance to UDCA. Participants were randomised in a 2:1 ratio stratified across two factors (ALP >3 x ULN or total bilirubin (TB) >ULN and PBC Worst Itch Numeric Rating Scale (WI-NRS) score ≥4) to receive elafibranor 80 mg or placebo once daily for at least 52 weeks. When applicable, participants continued their pre-study dose of UDCA throughout the study. Participants were included in the study if their ALP was ≥1.67 x ULN and TB was ≤2 x ULN. Participants were excluded in case of decompensated cirrhosis or other causes of liver disease.
Overall, the mean age was 57.1 years, and the mean weight was 70.8 kg. The study population was predominately female (96%) and white (91%). The baseline mean ALP concentration was 321.9 U/L, 39% of participants had a baseline ALP concentration >3 x ULN, and 35% of participants had advanced disease at baseline, defined as liver stiffness >10 kPa and/or bridging fibrosis or cirrhosis on histology.
The median duration of exposure was 63.07 and 61.00 weeks in the elafibranor and placebo groups, respectively.
The mean baseline TB concentration was 9.6 μmol/L and 96% of participants had a baseline TB concentration less than or equal to ULN. The mean baseline liver stiffness measurement by transient elastography was 10.1 kPa. The baseline mean PBC WI-NRS score was 3.3 and 41% had moderate-to- severe pruritus at baseline (PBC WI-NRS score ≥4); for those with moderate-to-severe pruritus, the baseline mean PBC WI-NRS score was 6.2 for participants in the elafibranor 80 mg group and 6.3 for participants in the placebo group. The majority (95%) of participants received treatment in combination with UDCA or as monotherapy in 5% of participants who were unable to tolerate UDCA.
The primary endpoint was cholestasis response at week 52 as defined as the composite endpoint: ALP <1.67 x ULN and TB ≤ ULN and ALP decrease ≥15%. The key secondary endpoints were ALP normalization at week 52 and the change in pruritus from baseline through week 52 and through week 24 based on the PBC WI-NRS score in participants with moderate-to-severe pruritus at baseline.
Table 1 shows the primary composite endpoint of cholestasis response and the key secondary endpoint of ALP normalization.
Table 1. Percentage of Adult Participants with PBC Achieving the Primary Efficacy Composite Endpoint of Cholestasis Response and Key Secondary Efficacy Endpoint of ALP Normalization at Week 52:
| Analysis Population | Elafibranor 80 mg (N=108) | Placebo (N=53) | Treatment Difference (95% CI)3 | Odds Ratio (95% CI)4 | P-value4 |
|---|---|---|---|---|---|
| Primary composite endpoint: Cholestasis response1 | |||||
| ITT | 51% | 4% | 47% (32, 57) | 37.6 (7.6, 302.2) | <0.0001 |
| First key secondary endpoint: ALP normalization2 | |||||
| ITT | 15% | 0 | 15% (6, 23) | Infinity (2.8, infinity) | 0.0019 |
ITT: Intention-to-treat
A significant decrease in ALP from baseline was seen as early as week 4 and was sustained over 52 weeks of treatment in the elafibranor group compared to placebo (Figure 1).
Figure 1. Mean (Least Squares (LS) mean with 95% CI) Change from Baseline in ALP Over Time - ITT analysis set:
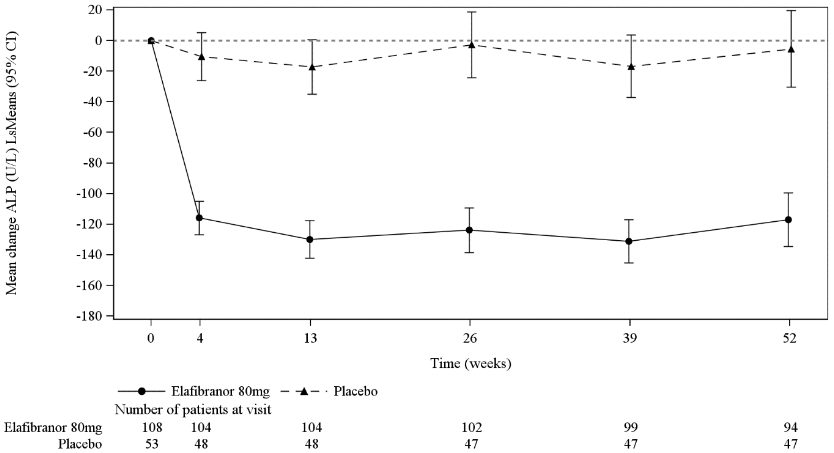
The primary endpoint of cholestasis response in participants with a baseline ALP ≤3 x ULN or TB <ULN was achieved in 71% of participants on elafibranor versus 6% of participants on placebo, compared to those with ALP >3 x ULN or TB >ULN where cholestasis response was achieved in 21% of participants on elafibranor versus 0% on placebo.
Among the 54 participants with advanced disease, 16/35 (46%) participants on elafibranor versus 0/19 (0%) participants on placebo achieved the primary endpoint of cholestasis response. Due to the limited number of participants with advanced disease, these results should be interpreted with caution.
Patient-reported outcomes
In participants with moderate-to-severe pruritus at baseline, a greater decrease from baseline in PBC WI-NRS score through week 52 and week 24 was observed in participants randomised to elafibranor compared to placebo but this did not reach statistical significance (Table 2).
Table 2. Change in Pruritus from Baseline Through Week 52 and Week 24 as Measured by PBC WI-NRS in those with Moderate-to-Severe Pruritus at Baseline:
| Elafibranor 80 mg (N=44) | Placebo (N=22) | Treatment Difference | P-value | |
|---|---|---|---|---|
| Second key secondary endpoint: change through week 521 | ||||
| Least Squares Mean (95% CI) | -1.9 (-2.6, -1.3) | -1.1 (-2.1, -0.2) | -0.8 (-2.0, 0.4) | 0.1970 |
| Third key secondary endpoint: change through week 241 | ||||
| Least Squares Mean (95% CI) | -1.6 (-2.2, -1.0) | -1.3 (-2.2, -0.3) | -0.3 (-1.5, 0.8) | - |
1 Analysis used the mixed model for repeated measures (MMRM) with treatment, 4-week period and treatment by 4-week period interaction as fixed factors and adjusting for baseline PBC WI-NRS and the stratification factor of ALP >3 x ULN or TB >ULN. An unstructured correlation structure is used. Treatment effect through week 52 is the average of NRS score changes from baseline for the thirteen 4-week periods. Treatment effect through week 52 and week 24 is the average treatment effects of NRS score changes from baseline over the first thirteen 4-week periods and first six 4-week periods, respectively. The assessments of PBC WI-NRS scores after participants stopped prematurely the study treatment or took a rescue therapy for pruritus were considered as missing.
Treatment with elafibranor was associated with an improvement in pruritus as evidenced by a reduction in the PBC-40 Itch and 5-D Itch total scores compared to placebo at week 52 (Table 3).
Table 3. Change in Pruritus from Baseline to Week 52 in PBC-40 Itch and 5-D Itch total scores in those with Moderate-to-Severe Pruritus at Baseline:
| Elafibranor 80 mg (N=44) | Placebo (N=22) | Treatment Difference | |
|---|---|---|---|
| PBC-40 Itch total score: change at week 521 | |||
| Least Squares Mean (95% CI) | -2.5 (-3.4, -1.6) | -0.1 (-1.6, 1.3) | -2.3 (-4.0, -0.7) |
| 5-D Itch total score: change at week 521 | |||
| Least Squares Mean (95% CI) | -4.2 (-5.6, -2.9) | -1.2 (-3.3, 0.9) | -3.0 (-5.5, -0.5) |
1 Analysis uses the mixed model for repeated measures (MMRM) with treatment, visits (until week 52) and treatment by visit interaction as fixed factor and adjusting for baseline score and the stratification factor of ALP > 3x ULN or TB > ULN.
Lipid parameters
Elafibranor demonstrated a favourable effect on lipid parameters. The mean reduction in very low-density lipoprotein-cholesterol (VLDL-C) and triglycerides (TG) was greater in participants treated with elafibranor compared to placebo at Week 52. The LS means difference from placebo in VLDL-C was -0.1 mmol/L [(95% CI: -0.2, -0.1); p<0.001] and for TG was -0.3 mmol/L [(95% CI: -0.4, -0.1)]; p<0.001]. High-density lipoprotein-cholesterol (HDL-C) remained stable on treatment with elafibranor.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Iqirvo in all subsets of the paediatric population in primary biliary cholangitis (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited.
The European Medicines Agency will review any new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Elafibranor plasma exposure (AUC) increases proportionally from 50 to 360 mg (0.6 to 4.5 times the recommended dose). Steady state is achieved by day 14 following once daily dosing. The pharmacokinetics (PK) of elafibranor and its major active metabolite GFT1007 was found to be time-independent after 16-day repeated administration. Elafibranor and its active metabolite exposure in participants with PBC are listed in Table 4.
Table 4. Elafibranor and GFT1007 exposures in participants with PBC at steady state following 80 mg QD (once daily):
| Cmax,ss (ng/mL) | AUC0-24 (ng • h/mL) | Accumulation ratio | |
|---|---|---|---|
| Elafibranor | 802 | 3758 | 2.9 |
| GFT1007 | 2058 | 11985 | 1.3 |
Absorption
Following repeated oral administration in participants with PBC, median peak plasma levels of elafibranor and GFT1007 at doses of 80 mg occur within 1.25 hours. When administered with a high-fat and high-calorie meal, there was a 30-minute delay in Tmax for elafibranor and a 1-hour delay for GFT1007 in fed compared to fasted conditions. The plasma exposure (AUC) of elafibranor decreased by 15% and the plasma AUC of GFT1007 was not affected. Given the higher circulating plasma levels of the pharmacologically active metabolite GFT1007 compared to elafibranor, food intake was deemed to have limited clinical impact based on overall exposure of parent and active metabolite.
Distribution
Plasma protein binding of both elafibranor and GFT1007 is approximately 99.7% (mainly to serum albumin). The mean apparent volume of distribution (Vd/F) of elafibranor in humans is 4731L, following single dose of elafibranor at 80 mg in fasted conditions.
Biotransformation
In vitro, elafibranor is metabolised by 15-ketoprostaglandin 13-Δ reductase (PTGR1). In vitro neither elafibranor nor GFT1007 show major metabolism by the main cytochrome P450 (CYP) and uridine diphosphate (UDP)-glucuronosyltransferase (UGT) isoforms. Following oral administration of 14C radiolabelled elafibranor, it was rapidly hydrolysed to the active metabolite GFT1007. Two major metabolites were identified in plasma, GFT1007 (active metabolite) and glucuronide conjugates (inactive metabolites).
Elimination
Following single 80 mg dose under fasted conditions, mean elimination half-life is 68.2 hours for elafibranor, and 15.4 hours for metabolite GFT1007. Elafibranor mean apparent total clearance (CL/F) was 50.0 L/h after a single 80 mg dose under fasted conditions.
Excretion
Following a single 120 mg oral dose of 14C radiolabelled elafibranor in healthy participants, approximately 77.1% of the dose was recovered in faeces, primarily as elafibranor (56.7% of the administered dose) and its active metabolite GFT1007 (6.08% of the administered dose). Approximately 19.3% recovered in urine, primarily as glucuronide conjugates.
Special populations
There was no evidence that age (from 18 to 80 years old), gender, race, Body Mass Index (BMI), and renal status, had any clinically meaningful impact on elafibranor and GFT1007 PK.
Hepatic impairment
The total drug exposure of the parent and active metabolite was not significantly different between participants with normal hepatic function and hepatically impaired participants (Child Pugh A, B and C). No dose adjustment is required for patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. However, the unbound fraction of elafibranor and GFT1007 increased by approximately 3-fold in the severe (Child Pugh C) hepatically impaired participants. Elafibranor is not recommended for patients with severe hepatic impairment (Child-Pugh C).
Drug-drug interactions
Based on in vitro studies, CYP and UGT enzymes were shown not to play a major role in elafibranor metabolism. Drug-drug interactions (DDI) are expected to be minimal with drugs that significantly alter CYP or UGT activity.
Clinical studies
Warfarin (CYP2C9 substrate):
Concomitant administration of elafibranor with warfarin resulted in no increase in exposure (AUC, Cmax) of warfarin, and no difference in international normalized ratio (INR) compared to warfarin alone.
Simvastatin (CYP3A, Breast Cancer Resistance Protein (BCRP), organic anion transporting polypeptides 1B1 (OATP1B1) and OATP1B3 substrates) and atorvastatin (CYP3A, organic anion transporting polypeptides 1B1 (OATP1B1) and OATP1B3 substrates):
Concomitant administration of repeat doses of elafibranor with simvastatin, or atorvastatin, resulted in no increase in exposure (AUC, Cmax) of simvastatin or its β-Hydroxyacid metabolite, or atorvastatin.
Sitagliptin (dipeptidyl peptidase-IV (DPP-IV) inhibitor):
No clinically significant effects on blood levels of GLP-1 were observed when co- administering 100 mg of elafibranor as a DDI perpetrator once daily for 15 days with a single oral 100 mg dose of sitagliptin during a meal test.
In Vitro Studies
Cytochrome P450 (CYP) inhibition and induction:
Elafibranor and GFT1007 were not considered inhibitors of main CYPs. No time dependent CYP inhibition was observed. Elafibranor and GFT1007 did not cause induction on CYP1A2, CYP2B6, and CYP3A4.
UGT inhibition:
Based on in vitro data elafibranor and GFT1007 were not expected to inhibit main UGTs at clinically significant concentrations.
Transporter systems:
Elafibranor was an inhibitor of OATP1B3 and BCRP. Based on the in vivo studies with simvastatin and atorvastatin, no clinical consequences are expected from the inhibition of OATP1B3 and BCRP.
5.3. Preclinical safety data
Nonclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeat dose toxicity, genotoxicity and carcinogenic potential.
Reproduction and development toxicity
Elafibranor has shown evidence of developmental toxicity in both rats and rabbits.In rat pre- and post-natal study, maternal exposures to elafibranor (at or above 2-fold the AUC exposure at the maximum human recommended dose (MHRD)) led to reduced pup survival, developmental delay, or thrombosis. In pregnant rabbits, maternal exposure (3-fold the AUC exposure at MHRD) to elafibranor caused marked maternal toxicity, increased embryo-lethality, reduced foetal weight and a low incidence of foetal malformations.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.