ITOVEBI Tablet Ref.[112407] Active ingredients: Inavolisib
Source: FDA, National Drug Code (US) Revision Year: 2024
12.1. Mechanism of Action
Inavolisib is an inhibitor of phosphatidylinositol 3-kinase (PI3K) with inhibitory activity predominantly against PI3Kα. In vitro, inavolisib induced the degradation of mutated PI3K catalytic alpha subunit p110α (encoded by the PIK3CA gene), inhibited phosphorylation of the downstream target AKT, reduced cellular proliferation, and induced apoptosis in PIK3CA-mutated breast cancer cell lines. In vivo, inavolisib reduced tumor growth in PIK3CA-mutated, estrogen receptor-positive, breast cancer xenograft models. The combination of inavolisib with palbociclib and fulvestrant increased tumor growth inhibition compared to each treatment alone or the doublet combinations.
12.2. Pharmacodynamics
Exposure-Response Relationships
The exposure-response relationship for the efficacy of inavolisib has not been fully characterized. Inavolisib time course of pharmacodynamic response is unknown. Higher systemic exposure of inavolisib was associated with higher incidence of Grade ≥2 anemia, Grade ≥2 hyperglycemia, and inavolisib dosage modifications due to adverse reactions.
Cardiac Electrophysiology
At the recommended approved dosage, a mean increase in the QTc interval of >20 ms is unlikely.
12.3. Pharmacokinetics
Inavolisib pharmacokinetics are presented as geometric mean (geometric coefficient of variation [geo CV]%) following administration of the approved recommended dosage unless otherwise specified. The inavolisib steady-state AUC is 1,010 h*ng/mL (25%) and Cmax is 69 ng/mL (27%). Steady-state concentrations are predicted to be attained by day 5.
Inavolisib accumulation is approximately 2-fold.
Inavolisib steady-state AUC is proportional with dose from 6 to 12 mg (0.7 to 1.3 times the approved recommended dosage).
Absorption
Inavolisib absolute oral bioavailability is 76%. Inavolisib steady-state median (min, max) time to maximum plasma concentration (Tmax) is 3 hours (0.5, 4 hours).
Effect of Food
No clinically significant differences in inavolisib pharmacokinetics were observed following administration of inavolisib with a high-fat meal (approximately 1,000 calories, 50% fat).
Distribution
Inavolisib apparent (oral) volume of distribution is 155 L (26%). Inavolisib plasma protein binding is 37% and is not concentration-dependent in vitro. Inavolisib blood-to-plasma ratio is 0.8.
Elimination
Inavolisib elimination half-life is 15 hours (24%) with a total clearance of 8.8 L/hr (29%).
Metabolism
Inavolisib is primarily metabolized by hydrolysis. In vitro, inavolisib was minimally metabolized by CYP3A.
Excretion
Following oral administration of a single radiolabeled dose, 49% of the administered dose was recovered in urine (40% unchanged) and 48% in feces (11% unchanged).
Specific Populations
No clinically significant differences in the pharmacokinetics of inavolisib were observed based on age (27 to 85 years), sex, race (Asian or White), body weight (39 to 159 kg), or mild hepatic impairment (total bilirubin > ULN to ≤ 1.5 × ULN or AST > ULN and total bilirubin ≤ ULN). The effect of moderate to severe hepatic impairment on inavolisib pharmacokinetics is unknown.
Patients with Renal Impairment
Inavolisib AUC was 73% higher in patients with moderate renal impairment compared to patients with normal renal function (eGFR ≥90 mL/min). No clinically significant differences in the pharmacokinetics of inavolisib were observed in patients with mild renal impairment compared to patients with normal renal function. The effect of severe renal impairment (eGFR <30 mL/min) on the pharmacokinetics of inavolisib is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Proton Pump Inhibitors: No clinically significant difference in steady-state inavolisib pharmacokinetics were observed based upon concomitant use of a proton pump inhibitor (lansoprazole, omeprazole, esomeprazole, pantoprazole, or rabeprazole).
In Vitro Studies
CYP450 Enzymes: Inavolisib induces CYP3A and CYP2B6. Inavolisib is a time-dependent inhibitor of CYP3A. Inavolisib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6.
Transporter Systems: Inavolisib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but is not a substrate of OATP1B1, OATP1B3, OCT1, OCT2, MATE1, MATE2K, OAT1, OAT2. Inavolisib does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, or MATE2K.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with inavolisib have not been conducted.
Inavolisib was not mutagenic in the bacterial reverse mutation (Ames) assay. Inavolisib was clastogenic in an in vitro human lymphocyte micronucleus assay. Inavolisib was not genotoxic in an in vivo rat bone marrow micronucleus test and did not induce DNA break in a liver comet assay.
Fertility studies with inavolisib have not been conducted. In repeat-dose toxicity studies, inavolisib was administered orally once daily for up to 3 months duration in rats and dogs.
In male rats, dose-dependent atrophy of the prostate and seminal vesicle and decreased organ weights of the prostate, seminal vesicle, epididymis and testis were observed at doses ≥1.5 mg/kg/day (≥0.4 times the human exposure at the recommended dose of 9 mg/day based on AUC). In male dogs, focal inspissation of seminiferous tubule contents and multinucleated spermatids in the testis and epithelial degeneration/necrosis in the epididymis were observed following 4 weeks of dosing at ≥1.5 mg/kg/day (≥2 times the human exposure at the recommended dose of 9 mg/day based on AUC) and decreased sperm count was observed following 3 months of dosing at ≥1 mg/kg/day (≥1.2 times the human exposure at the recommended dose of 9 mg/day based on AUC). Findings in dogs were not observed following a recovery period.
In female rats, atrophy in the uterus and vagina, decreased ovarian follicles, and findings suggestive of an interruption/alteration of the estrous cycle were observed following up to 3 months of dosing at doses ≥3 mg/kg/day (≥1.2 times the human exposure at the recommended dose of 9 mg/day based on AUC). Findings in the uterus, vagina, and estrous cycle observed in the 4-week toxicity study were not observed following recovery. Recovery was not assessed in the 3-month study in rats.
13.2. Animal Toxicology and/or Pharmacology
Lens degeneration, characterized by lens fiber swelling, separation of lens fibers, and/or accumulation of subcapsular proteinaceous material, was observed in rats at an oral inavolisib dose of 10 mg/kg/day (6.3 times the human exposure at the recommended dose of 9 mg/day based on AUC). In dogs, lens fiber swelling and lens cortex vacuolation were observed at oral inavolisib doses ≥0.3 mg/kg/day (≥0.5 times the human exposure at the recommended dose based on AUC) and ≥1 mg/kg/day (≥1.2 times the human exposure at the recommended dose based on AUC), respectively. Lens degeneration was present in rats following a 4-week recovery period but was not present in dogs following a 12-week recovery period.
14. Clinical Studies
14.1 Locally Advanced or Metastatic Breast Cancer
INAVO120
INAVO120 (NCT04191499) was a randomized (1:1), double-blind, placebo-controlled trial evaluating the efficacy of ITOVEBI in combination with palbociclib and fulvestrant in adult patients with endocrine-resistant PIK3CA-mutated, HR-positive, HER2-negative (defined as IHC 0 or 1+, or IHC 2+/ISH-), locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease. Randomization was stratified by presence of visceral disease (yes or no), endocrine resistance (primary or secondary), and geographic region (North America/Western Europe, Asia, other).
Primary endocrine resistance was defined as relapse while on the first 2 years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least 2 years or relapse within 12 months of completing adjuvant ET.
Patients were required to have HbA1C < 6% and fasting blood glucose < 126 mg/dL. The study excluded patients with Type 1 diabetes mellitus or Type 2 diabetes mellitus requiring ongoing anti-hyperglycemic treatment at the start of study treatment.
PIK3CA mutation status was prospectively determined in a central laboratory using the FoundationOne Liquid CDx assay on plasma-derived circulating tumor DNA (ctDNA) or in local laboratories using various validated polymerase chain reaction (PCR) or next-generation sequencing (NGS) assays on tumor tissue or plasma. All patients were required to provide both a freshly collected pre-treatment blood sample and a tumor tissue sample for central evaluation and determination of PIK3CA mutation(s) status.
Patients received either ITOVEBI 9 mg (n=161) or placebo (n=164) orally once daily, in combination with palbociclib 125 mg orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a cycle of 28 days, and fulvestrant 500 mg administered intramuscularly on Cycle 1, Days 1 and 15, and then on Day 1 of every 28-day cycle. Patients received treatment until disease progression or unacceptable toxicity. In addition, all pre/perimenopausal women and men received an LHRH agonist throughout therapy.
The baseline demographic and disease characteristics were: median age 54 years (range: 27 to 79 years); 98% female, of which 39% were pre/perimenopausal; 59% White, 38% Asian, 2.5% unknown, 0.6% Black or African American; 6% Hispanic or Latino; and Eastern Cooperative Oncology Group (ECOG) performance status of 0 (63%) or 1 (36%). Tamoxifen (57%) and aromatase inhibitors (50%) were the most commonly used adjuvant endocrine therapies. Sixty-four percent of patients were considered to have secondary endocrine resistance. Eighty-three percent of patients had received prior chemotherapy (in the neo/adjuvant setting) and 1.2% of patients had been treated with a CDK4/6 inhibitor.
The major efficacy outcome measure was investigator (INV)-assessed progression-free survival (PFS) per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Additional efficacy outcome measures included overall survival (OS), INV-assessed objective response rate (ORR), and INV-assessed duration of response (DOR).
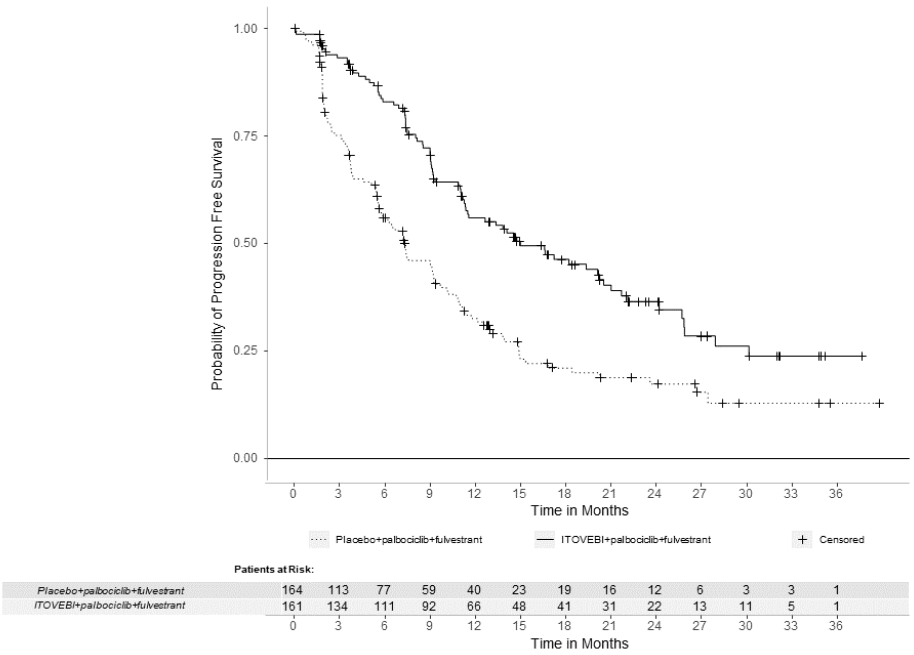
Efficacy results are summarized in Table 6 and Figure 1. INV-assessed PFS results were supported by consistent results from a blinded independent central review (BICR) assessment. At the time of the PFS analysis, OS data were not mature with 30% deaths in the overall population.
Table 6. Efficacy Results in Patients with Locally Advanced or Metastatic Breast Cancer in INAVO120:
| Efficacy Endpoint | ITOVEBI + Palbociclib + Fulvestrant N=161 | Placebo + Palbociclib + Fulvestrant N=164 |
|---|---|---|
| Progression-Free Survivala,b | ||
| Patients with event, n (%) | 82 (51) | 113 (69) |
| Median, months (95% CI) | 15.0 (11.3, 20.5) | 7.3 (5.6, 9.3) |
| Hazard ratio (95% CI) | 0.43 (0.32, 0.59) | |
| p-value | <0.0001 | |
| Objective Response Ratea,b,c | ||
| Patients with CR or PR, n (%) | 94 (58) | 41 (25) |
| 95% CI | (50, 66) | (19, 32) |
| Duration of Responseb | ||
| Median DOR, months (95% CI) | 18.4 (10.4, 22.2) | 9.6 (7.4, 16.6) |
CI = confidence interval; CR = complete response; DOR = duration of response; PR = partial response
a Per RECIST version 1.1.
b Based on investigator assessment.
c Based on confirmed ORR.
Figure 1. Kaplan-Meier Curve for Investigator-Assessed Progression-Free Survival in INAVO120:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.