JEMPERLI Concentrate for solution for infusion Ref.[27949] Active ingredients: Dostarlimab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: GlaxoSmithKline (Ireland) Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Anti-neoplastic agents, monoclonal antibodies and antibody drug conjugates
ATC code: L01FF07
Mechanism of action
Dostarlimab is a humanised mAb of the IgG4 isotype that binds to PD-1 receptors and blocks the interactions of binding with its ligands PD-L1 and PD-L2. The inhibition of PD-1 pathway-mediated immune response results in reactivation of T-cell function such as proliferation, cytokine production, and cytotoxic activity. Dostarlimab potentiates T-cell responses, including anti-tumour immuno responses through blockade of PD-1 binding to PD-L1 and PD-L2. In syngeneic mouse tumour models, blocking PD-1 activity resulted in decreased tumour growth.
Clinical efficacy and safety
RUBY: Randomised controlled study of dostarlimab in combination with carboplatin and paclitaxel in treatment of adult patients with primary advanced or recurrent EC
The efficacy and safety of dostarlimab in combination with carboplatin-paclitaxel were investigated in a multicentre, randomised, double blinded, placebo-controlled Phase 3 study conducted in patients with primary advanced or recurrent EC.
Patients were randomised (1:1) to receive dostarlimab 500 mg plus carboplatin AUC 5 mg/mL/min and paclitaxel 175 mg/m² every 3 weeks for 6 cycles followed by dostarlimab 1000 mg every 6 weeks (n=245) or placebo plus carboplatin AUC 5 mg/mL/min and paclitaxel 175 mg/m² every 3 weeks for 6 cycles followed by placebo every 6 weeks (n=249). Randomisation was stratified by MMR/MSI status, prior external pelvic radiotherapy, and disease status (recurrent, primary Stage III, or primary Stage IV). Treatment continued for up to 3 years or until unacceptable toxicity, disease progression or investigator decision. Assessment of tumour status was performed every 6 weeks through week 25, every 9 weeks through week 52 and every 12 weeks thereafter. After a median follow-up of 30 months, 6 out of 53 patients randomised to dostarlimab plus carboplatin-paclitaxel have received treatment for >3 years (cutoff date 01 Mar 2023).
The key eligibility criteria for the study were International Federation of Gynaecology and Obstetrics (FIGO) primary Stage III or Stage IV disease, including Stage IIIA to IIIC1 disease with presence of evaluable or measurable disease per RECIST v.1.1, Stage IIIC1 patients with carcinosarcoma, clear cell, serous, or mixed histology (containing ≥10% carcinosarcoma, clear cell, or serous histology) regardless of presence of evaluable or measurable disease on imaging, Stage IIIC2 or Stage IV disease regardless of presence of evaluable or measurable disease. The study also included patients with first recurrent EC with a low potential for cure by radiation therapy or surgery alone or in combination, including patients who had first recurrent disease and were naïve to systemic anticancer therapy or who had received prior neoadjuvant/adjuvant systemic anticancer therapy and had a recurrence or progressive disease ≥6 months after completing treatment (first recurrence). Prior radiation was not permitted within 21 days of study treatment excluding palliative radiotherapy which was permitted within up to 1 week of study treatment.
The primary efficacy outcome measures were progression-free survival (PFS) assessed by the investigator according to RECIST v1.1 in subjects with dMMR/MSI-H primary advanced or recurrent EC and in all subjects (overall ITT population) with primary advanced or recurrent EC, and overall survival (OS) in all subjects (overall ITT population) with primary advanced or recurrent EC.
A total of 118 patients with dMMR/MSI-H EC were evaluated for efficacy in the RUBY study. Baseline demographics and characteristics were: median age 64 years (34% aged 65 to 74 years and 15% aged 75 years or older); 85% White, 9% Black, 2% Asian; ECOG PS 0 (57%) or 1 (43%); primary stage III 21%, primary stage IV 30%, recurrent EC 49%; endometrioid carcinoma 85%, mixed carcinoma 5%, carcinosarcoma 4%, serous carcinoma 2%, other 4%; and prior surgery 92%, prior radiotherapy (35%), prior anti-cancer therapy (14%).
The identification of dMMR/MSI-H tumour status was prospectively determined based on local testing assays (IHC, PCR or NGS), or central testing (IHC) when no local result was available.
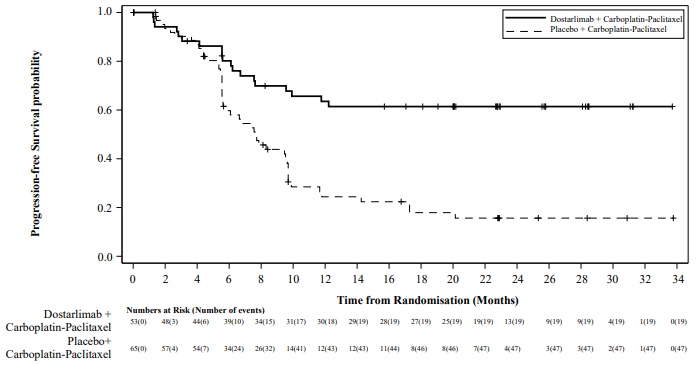
Efficacy results are shown in Table 5 and Figure 1. All endpoints are presented at the primary analysis for PFS with median follow up of 25 months. OS results are based on the first OS interim analysis. The RUBY study demonstrated a statistically significant improvement in PFS by investigator in patients randomised to dostarlimab plus carboplatin-paclitaxel versus placebo plus carboplatin-paclitaxel.
Table 5. Efficacy results in RUBY for patients with dMMR/MSI-H EC:
| Endpoint | Dostarlimab + carboplatin-paclitaxel (N=53)a | Placebo + carboplatin-paclitaxel (N=65)a |
|---|---|---|
| Progression free-survival (PFS) | ||
| Median in months (95% CI)b | Not reached | 7.7 (5.6, 9.7) |
| Number (%) of patients with event | 19 (35.8) | 47 (72.3) |
| Hazard ratio (95% CI)c | 0.28 (0.16, 0.50) | |
| p-valueb | <0.0001 | |
| Overall survival (OS)d | ||
| Median in months | Not reached | Not reached |
| Number (%) of patients with event | 7 (13.2) | 24 (36.9) |
| Hazard ratio (95% CI)c | 0.30 (0.13, 0.70) | |
CI: Confidence interval
a Efficacy data with a median follow-up of 25 months (cut-off date 28 Sept 2022).
b One-sided p-value based on stratified log-rank test.
c Based on stratified Cox regression model.
d Not statistically significant since no hypothesis testing was performed for overall survival in the dMMR/MSI-H population.
Figure 1. Kaplan-Meier curve of progression-free survival per investigator assessment in patients with dMMR/MSI-H EC (RUBY study):
GARNET: adult patients with recurrent or advanced dMMR/MSI-H EC who have progressed on or after treatment with a platinum-containing regimen
The efficacy and safety of dostarlimab monotherapy were investigated in the GARNET study, a multicentre, uncontrolled, multiple parallel cohort, open-label study. The GARNET study included expansion cohorts in subjects with recurrent or advanced solid tumours who have limited available treatment options. Cohort A1 enrolled patients with dMMR/MSI-H EC who have progressed on or after a platinum-containing regimen.
Patients received 500 mg dostarlimab every 3 weeks for 4 cycles followed by 1000 mg dostarlimab every 6 weeks. Treatment continued until unacceptable toxicity or disease progression for up to two years.
The major efficacy outcome measures were objective response rate (ORR) and duration of response (DOR) as assessed by blinded independent central radiologists' (BICR) review according to response evaluation criteria in solid tumours (RECIST) v 1.1. The efficacy population was defined as patients who had measurable disease by BICR at baseline and had minimum of 24 weeks follow-up or had less than 24 weeks of follow-up and discontinued due to adverse events or disease progression.
A total of 143 patients with dMMR/MSI-H EC were evaluated for efficacy in the GARNET study. Among these 143 patients, the baseline characteristics were: median age of 65 years (52% aged 65 years or older); 77% white, 3.5% Asian, 2.8% black; and ECOG PS 0 (39%) or 1 (61%). At the time of diagnosis, 21% of the patients with dMMR/MSI-H EC were FIGO Stage IV. At study entry (the most recent FIGO stage), 67% of the patients were FIGO Stage IV. The median number of prior lines of herapy was one: 63% of patients had one prior line, 37% had two or more prior lines. Forty-nine patients (34%) received treatment only in the neoadjuvant or adjuvant setting before participating in the study.
The identification of dMMR/MSI-H tumour status was prospectively determined based on local testing. Local diagnostic assays (IHC, PCR or NGS) available at the sites were used for the detection of the dMMR/MSI-H expression in tumour material. Most of the sites used IHC as it was the most common assay available.
Table 6 includes the efficacy data for the 143 patients. The overall median treatment duration in weeks was 34 (range 2 to 220). Twenty four percent of subjects who received any amount of dostarlimab received treatment >102 weeks (2 years).
Table 6. Efficacy results in GARNET for patients with dMMR/MSI-H EC:
| Endpoint | Results (N=143)a |
|---|---|
| Objective response rate (ORR) | |
| ORR n (%) (95% CI) | 65 (45.5) (37.1, 54.0) |
| Complete response rate, n (%) | 23 (16.1) |
| Partial response rate, n (%) | 42 (29.4) |
| Duration of response (DOR)b | |
| Median in months | Not reached |
| Patients with duration ≥12 months, n (%) | 52 (80.0) |
| Patients with duration ≥24 months, n (%) | 29 (44.6) |
| Disease control rate (DCR)c | |
| DCR n (%) (95% CI) | 86 (60.1) (51.6, 68.2) |
CI: Confidence interval
a Efficacy data with a median follow-up of 27.6 months (cut-off date 01 Nov 2021)
b For patients with a partial or complete response.
c Includes patient with complete response, partial response and stable disease for at least 12 weeks.
Efficacy and PD-L1 status
Clinical activity was observed regardless of tumour PD-L1 combined positive score (CPS) by IHC. The relationship between PD-L1 status and efficacy was analysed post-hoc in patients with available tissue samples (N=81) among the efficacy population from Cohort A1 of the GARNET study using a data cutoff date of 01 March 2020. Among 23 patients with PD-L1 CPS <1%, ORR was 30.4% (7/23, 95% CI 13.2, 52.9) and among 58 patients with PD-L1 CPS ≥1%, ORR was 55.2% (32/58, 95% CI 41.5, 68.3).
Elderly patients
Of the 108 patients treated with dostarlimab in the GARNET study efficacy population, 50.0% were older than 65 years. Consistent results were observed in the elderly population, where the ORR by BICR (95% CI) was 42.6% (29.2%, 56.8%) in patients ≥65 years.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with dostarlimab in all subsets of the paediatric population in the treatment of all conditions included in the category of malignant neoplasms, except haematopoietic and lymphoid tissue (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics (PK) of dostarlimab were assessed as a monotherapy and when administered in combination with carboplatin and paclitaxel.
Dostarlimab monotherapy or in combination with carboplatin and paclitaxel was characterised using population PK analysis from 869 patients with various solid tumours, including 546 patients with EC. When dosed at the recommended therapeutic dose for monotherapy (500 mg administered intravenously every 3 weeks for 4 doses, followed by 1000 mg every 6 weeks), or at the recommended therapeutic dose for combination with carboplatin and paclitaxel (500 mg administered intravenously every 3 weeks for 6 doses, followed by 1000 mg every 6 weeks), dostarlimab shows an approximate two-fold accumulation (Cmin), consistent with the terminal half-life (t1/2). The exposure of dostarlimab as monotherapy and/or in combination with carboplatin and paclitaxel was similar.
Absorption
Dostarlimab is administered via the intravenous route and therefore estimates of absorption are not applicable.
Distribution
The mean volume of distribution of dostarlimab at steady state is approximately 5.8 L (CV % of 14.9%).
Biotransformation
Dostarlimab is a therapeutic mAb IgG4 that is expected to be catabolised into small peptides, amino acids, and small carbohydrates by lysosome through fluid-phase or receptor-mediated endocytosis. The degradation products are eliminated by renal excretion or returned to the nutrient pool without biological effects.
Elimination
The mean clearance is 0.007 L/h (CV % of 30.2%) at steady state. The t1/2 at steady state is 23.2 days (CV % of 20.8%).
Dostarlimab clearance was estimated to be 7.8% lower when dostarlimab was given in combination with carboplatin and paclitaxel. There was no meaningful impact on dostarlimab exposure.
Linearity/non-linearity
Exposure (both maximum concentration [Cmax] and the area under the concentration-time curve, [AUC0-tau] and [AUC0-inf]) was approximately dose proportional.
Pharmacokinetic/pharmacodynamic relationship
Based on exposure efficacy and safety relationships, there are no clinically significant differences in efficacy and safety when doubling the exposure of dostarlimab. Full receptor occupancy as measured by both the direct PD-1 binding and interleukin 2 (IL-2) production functional assay was maintained throughout the dosing interval at the recommended therapeutic dosing regimen.
Special populations
A population PK analysis of the patient data indicates that there are no clinically important effects of age (range: 24 to 86 years), gender or race, ethnicity, or tumour type on the clearance of dostarlimab.
Renal impairment
Renal impairment was evaluated based on the estimated creatinine clearance [CLCR mL/min] (normal: CLCR ≥ 90 mL/min, n=305; mild: CLCR = 60-89 mL/min, n=397; moderate: CLCR = 30-59 mL/min, n=164; severe: CLCR = 15-29 mL/min, n=3 and ESRD: CLCR < 15 mL/min, n=1). The effect of renal impairment on the clearance of dostarlimab was evaluated by population pharmacokinetic analyses in patients with mild or moderate renal impairment compared to patients with normal renal function. No clinically important differences in the clearance of dostarlimab were found between patients with mild or moderate renal impairment and patients with normal renal function. There are limited data in patients with severe renal impairment.
Hepatic impairment
Hepatic impairment was evaluated as defined using the US National Cancer Institute criteria of hepatic dysfunction by total bilirubin and AST (Normal: total bilirubin (TB) & AST < = upper limit of normal (ULN), n=772; mild: TB > ULN to 1.5 ULN or AST > ULN, n=92; and moderate: TB > 1.5-3 ULN, any AST, n=5). The effect of hepatic impairment on the clearance of dostarlimab was evaluated by population pharmacokinetic analyses in patients with mild hepatic impairment compared to patients with normal hepatic function. No clinically important differences in the clearance of dostarlimab were found between patients with mild hepatic impairment and normal hepatic function. There are limited data in patients with moderate hepatic impairment and no data in patients with severe hepatic impairment.
5.3. Preclinical safety data
Nonclinical data reveal no special hazard for humans based on repeat-dose toxicity studies of duration up to 3 months in the cynomolgus monkey. No studies have been performed to assess the potential of dostarlimab for carcinogenicity or genotoxicity. Animal reproduction and development toxicity studies have not been conducted with dostarlimab. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to result in an increase in foetal loss. These results indicate a potential risk that administration of dostarlimab during pregnancy could cause foetal harm, including increased rates of abortion or stillbirth.
No notable effects on the male and female reproductive organs were observed in monkeys in the 1-month and 3-month repeat-dose toxicology studies; however, these results may not be representative at all of the potential clinical risk because of the immaturity of the reproductive system of animals used in the studies. Therefore, fertility toxicity remains unknown.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.