JERAYGO Film-coated tablet Ref.[111534] Active ingredients: Aprocitentan
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Idorsia Pharmaceuticals Deutschland GmbH, Marie-Curie-Strasse 8, 79539 Lörrach, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antihypertensives, other antihypertensives
ATC code: C02KN01
Mechanism of action
Endothelin (ET)-1, via its receptors (ETA and ETB), mediates a variety of effects such as vasoconstriction, fibrosis, cell proliferation, and inflammation and is upregulated in hypertension. Aprocitentan is a dual ERA that inhibits the binding of ET-1 to ETA and ETB receptors and hence the effects mediated by these receptors.
Pharmacodynamic effects
Cardiac electrophysiology
In a thorough QT study in healthy subjects, once-daily administration of 25 mg (maximum therapeutic dose) aprocitentan at steady state did not prolong the QTc interval as the upper limit of the 90% confidence interval of the mean change from baseline in placebo-corrected QTc was less than 10 ms.
At four times the maximum therapeutic dose (100 mg), the upper limit of the 90% confidence interval of the mean change from baseline in placebo-corrected QTc was 10.4 ms.
Clinical efficacy and safety
The efficacy of aprocitentan was evaluated in one randomized, double-blind, placebo-controlled Phase 3 multicentre study.
Patients with uncontrolled BP (systolic blood pressure [SBP] ≥140 mmHg) despite the use of at least three antihypertensive medicinal products and following exclusion of pseudo-resistant hypertension (e.g., white coat effect, inappropriate BP measurement, secondary causes of hypertension) were considered to have resistant hypertension.
The patients were switched to standardised background antihypertensive therapy consisting of an angiotensin receptor blocker (valsartan 160 mg), a calcium channel blocker (amlodipine 5 or 10 mg), and a diuretic (hydrochlorothiazide 25 mg) throughout the study. Patients with concomitant use of beta-blockers continued this treatment throughout the study, in addition to the standardised background antihypertensive therapy and study treatment.
A total of 730 patients received either aprocitentan 12.5 mg, aprocitentan 25 mg, or placebo once daily during the initial 4-week DB treatment (part 1). Thereafter, patients received aprocitentan 25 mg once daily during the 32-week SB treatment (part 2). At the end of the 32 weeks, patients were re-randomised to receive either aprocitentan 25 mg or placebo, once daily, during the 12-week DB-WD treatment (part 3) (Table 2).
Table 2. Design of the Phase 3 study:
| Treatment | Part 1 (4 weeks) | Part 2 (32 weeks) | Part 3 (12 weeks) | |
|---|---|---|---|---|
| Design | DB, placebo- controlled, randomized (1:1:1) | SB | DB-WD, placebo- controlled, randomized (1:1) | |
| Duration | Week 0 – Week 4 | Week 4 – Week 36 | Week 36 – Week 48 | |
| Treatment as add-on to background therapy* | Aprocitentan 25 mg Aprocitentan 12.5 mg Placebo | N=243 N=243 N=244 | N=704 | N=307 N=307 |
* ARB, CCB, and a diuretic.
ARB = angiotensin receptor blocker; CCB = calcium channel blocker; DB = double-blind; DB-WD = double-blind withdrawal; N = number of patients; SB = single-blind.
The primary efficacy endpoint was the change in sitting SBP (SiSBP) from baseline to Week 4 during DB treatment (part 1), measured at trough by unattended automated office blood pressure (uAOBP).
The key secondary endpoint was the change in SiSBP measured at trough by uAOBP from DB-WD baseline (Week 36) to Week 40 (part 3).
Patients had a mean age of 61.7 years (range 24 to 84 years; 34.1% were ≥65 and <75 years; 9.9% were ≥75 years) and 59.5% were male. Patients were White (82.9%), African American (11.2%) or Asian (5.2%). The mean body weight was 97.6 kg (range 46 to 196 kg) and mean BMI was 33.7 kg/m² (range 18 to 64 kg/m²).
Patients had a medical history of type 2 diabetes mellitus (54.1%), ischaemic heart disease (30.8%), central nervous system vascular disorders (23.0%), chronic kidney disease stages 3 and 4 (22.2%; 19.3% of patients had eGFR 30–59 mL/min/1.73 m² and 2.9% had eGFR 15–29 mL/min/1.73 m²), congestive heart failure (19.6%), and sleep apnoea syndrome (14.1%). 63.0% of patients had four or more antihypertensive medicinal products.
Populations not studied in the Phase 3 study are described in sections 4.2, 4.3 and 4.4.
Doses of aprocitentan 12.5 and 25 mg showed a statistically significant reduction vs placebo on SiSBP at Week 4. The treatment effect was consistent for sitting diastolic BP (SiDBP) (Table 3).
Table 3. Reduction in sitting trough BP (mmHg) measured by uAOBP at Week 4 of DB treatment:
| Difference to placebo | |||||
|---|---|---|---|---|---|
| Treatment group | N | Baseline# Mean | LS Mean | LS Mean | p-value |
| SiSBP (primary endpoint) | LS Mean (97.5% CL) | LS Mean (97.5% CL) | |||
| 12.5 mg | 243 | 153.2 | −15.3 (−17.4, −13.2) | −3.8 (−6.8, −0.8) | 0.0042* |
| 25 mg | 243 | 153.3 | −15.2 (−17.3, −13.1) | −3.7 (−6.7, −0.8) | 0.0046* |
| Placebo | 244 | 153.3 | −11.5 (−13.6, −9.4) | - | - |
| SiDBP | LS Mean (95% CL) | LS Mean (95% CL) | |||
| 12.5 mg | 243 | 87.9 | −10.4 (−11.6, −9.3) | −3.9 (−5.6, −2.3) | <0.0001 |
| 25 mg | 243 | 87.7 | −11.0 (−12.1, −9.8) | −4.5 (−6.1, −2.9) | <0.0001 |
| Placebo | 244 | 87.1 | −6.5 (−7.6, −5.3) | - | - |
# Observed baseline value.
* Statistically significant at the 2.5% level as prespecified in the testing strategy.
CL = confidence limit; DB = double-blind; DB-WD = double-blind withdrawal; LS Mean = least squares mean; SiDBP = sitting diastolic blood pressure; SiSBP = sitting systolic blood pressure.
The persistence of the BP-lowering effect of aprocitentan was shown in DB-WD treatment (part 3). In patients re-randomised to placebo, the mean SiSBP increased, whereas in patients re-randomised to aprocitentan 25 mg the mean effect on SiSBP was stable, resulting in a statistically significant difference. The treatment effect was consistent for SiDBP (Table 4).
Table 4. Sustained reduction in sitting trough BP (mmHg) measured by uAOBP at Week 40 of DB-WD treatment:
| Difference to placebo | |||||
|---|---|---|---|---|---|
| Treatment group | N | DB-WD Baseline# Mean | LS Mean (95% CL) | LS Mean (95% CL) | p-value |
| SiSBP (key secondary endpoint) | |||||
| 25 mg | 307 | 135.3 | −1.5 (−3.0, 0.0) | −5.8 (−7.9, −3.7) | <0.0001* |
| Placebo | 307 | 136.4 | 4.4 (2.9, 5.8) | - | - |
| SiDBP | |||||
| 25 mg | 307 | 76.1 | −0.5 (−1.5, 0.5) | −5.2 (−6.6, −3.8) | <0.0001 |
| Placebo | 307 | 76.3 | 4.7 (3.7, 5.7) | - | - |
# Observed baseline value. DB-WD baseline: Week 36.
* Statistically significant at the 5% level as prespecified in the testing strategy.
CL = confidence limit; DB-WD = double-blind-withdrawal; LS Mean = least squares mean; SiDBP = sitting diastolic blood pressure; SiSBP = sitting systolic blood pressure.
The effect was also consistent across SBP and DBP measured by ambulatory BP monitoring (ABPM) and assessed as daytime, night-time, and 24 h periods at Week 4 (Figure 1) and Week 40.
Figure 1. Placebo-corrected changes from baseline in systolic and diastolic BP measured by ABPM at Week 4:
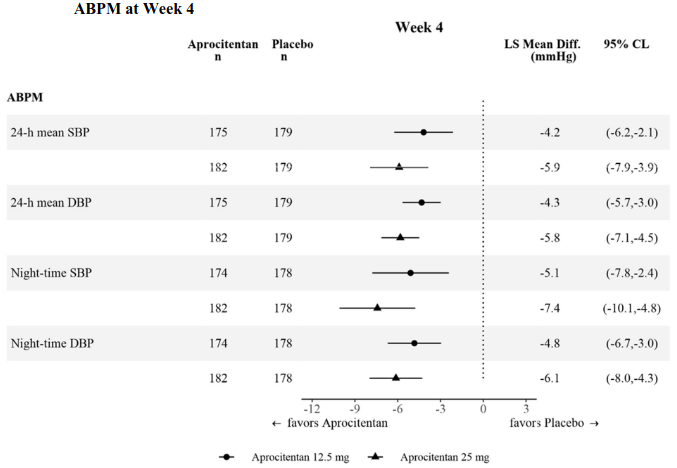
ABPM = ambulatory blood pressure monitoring; BP = blood pressure; CL = confidence limits; DBP = diastolic blood pressure; LS Mean Diff. = least squares mean difference versus placebo; SBP = systolic blood pressure.
A substantial proportion (i.e., at least 90%) of the BP-lowering effect was observed within the first two weeks of treatment with aprocitentan.
Figure 2. Mean sitting systolic BP measured by uAOBP over 48 weeks:
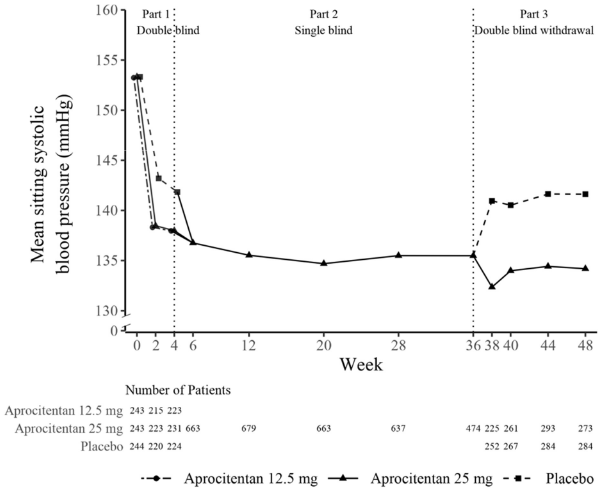
The effect of aprocitentan was consistent across subgroups of age (including patients ≥75 years), sex, race (including patients with Black or African American origin), BMI, baseline urine albumin-to-creatinine ratio (UACR), baseline eGFR and medical history of diabetes, and was consistent with the effect in the overall population.
Effects on UACR/eGFR
At 4 weeks, a reduction in UACR of 30% (95% confidence limits 20–39%) and 34% (95% confidence limits 25–42%) was observed with aprocitentan 12.5 and 25 mg, respectively, compared to subjects randomised to placebo. This effect disappeared upon treatment discontinuation. As for eGFR, a mean decrease of −1.2 mL/min /1.73 m² for aprocitentan 12.5 mg and −2.4 mL/min /1.73 m² for aprocitentan 25 mg occurred during the first 4 weeks of treatment (vs −0.6 mL/min/1.73 m² for placebo), followed by a stabilisation of eGFR, including in patients with low (<60 mL/min) baseline values, until the end of the study. The effect of aprocitentan on end organ protection has not been studied.
Effects on mortality and cardiovascular morbidity
The effects of aprocitentan on mortality and cardiovascular morbidity have not been studied.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with aprocitentan in all subsets of the paediatric population in the treatment of hypertension (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Maximum plasma concentration (Cmax) of aprocitentan was achieved between 4 and 5 h after administration of 25 mg. Concentrations in plasma increased in a dose-proportional manner following once daily administration of 5 mg, 25 mg, and 100 mg. The absolute bioavailability after oral administration is not known.
With once daily administration, steady-state conditions were reached by Day 8 and accumulation compared to Day 1 was approximately 3-fold.
Effect of food
When a capsule formulation (used in early clinical studies) was taken with a high-fat, high-calorie meal by healthy subjects, aprocitentan median time to Cmax (tmax) was reached approximately one hour earlier, with a Cmax approximately 1.7-fold that in the fasted condition. Total exposure expressed as AUC0-∞ was approximately 1.2-fold that observed in the fasted condition. Food effect has not been specifically studied for the film-coated tablet. In the pivotal Phase 3 study, aprocitentan film-coated tablets were administered irrespective of food intake. The absorption of aprocitentan is not expected to be affected by meals.
Distribution
Aprocitentan had an apparent volume of distribution of approximately 20 L and was highly bound to plasma proteins (>99%). The blood-to-plasma ratio was 0.63.
Biotransformation
Aprocitentan was almost exclusively detected unchanged in plasma. The main metabolic pathways of aprocitentan were N-glucosidation of the sulfamide moiety catalysed by the glucuronyl transferases UGT1A1 and UGT2B7, and hydrolysis of the sulfamide moiety to the corresponding aminopyrimidine. Hydrolysis was mostly non-enzymatic.
Elimination
After administration of a radiolabelled dose of aprocitentan, approximately 52% of radioactive drug-related material was eliminated via urine and 25% via faeces. A total of 0.2% and 6.8% of the administered dose was recovered in urine and faeces as unchanged aprocitentan, respectively.
The apparent oral body clearance is 0.30 L/h. The terminal plasma half-life of aprocitentan is approximately 46 h.
Pharmacokinetics in special populations
There were no clinically relevant effects of age (18–84 years), sex, body weight, or race on the PK of aprocitentan.
Renal impairment
Total exposure to aprocitentan (AUC) in patients with severe renal impairment (eGFR 15–29 mL/min) compared to healthy subjects was increased by an average of 40%. This increase is not considered clinically relevant (see section 4.2). Aprocitentan binding to plasma proteins was not influenced by renal function.
Hepatic impairment
Total exposure to aprocitentan (AUC) in patients with moderate hepatic impairment (Child-Pugh class B) compared to healthy subjects was increased by an average of 23%. This increase is not considered clinically relevant (see section 4.2). Aprocitentan binding to plasma proteins was not influenced by hepatic function.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated-dose toxicity, genotoxicity, carcinogenic potential, and phototoxicity.
Histological findings in repeated-dose toxicity studies (degenerative liver changes, nasal cavity findings, and testicular changes) were observed only at exposures sufficiently in excess of the maximum human exposure, indicating low relevance in clinical use.
Toxicity to reproduction and development
Testicular tubular degeneration was observed after repeated dosing in rats and dogs with safety margins of 8 (20.6)- and 4.9 (16.6)-fold the total (free) exposure at the maximum recommended human dose, respectively. However, no effects were noted on fertility or spermatogenesis in male rats.
In female rats, minimally increased pre-implantation loss (lower number of corpora lutea, implantation sites, and live embryos) was observed at 11 (29)-fold the total (free) exposure at the maximum recommended human dose. No effects on mating behaviour and reproductive performance were noted.
Aprocitentan did not induce teratogenicity in studies with pregnant rats and rabbits with safety margins of 2 (6)- and 14 (3)-fold the total (free) exposure at the maximum recommended human dose, respectively. However, ERAs as a class have shown teratogenicity in rats and rabbits, where the observed malformations indicate serious effects on developmental processes early in pregnancy (neural crest cell migration). Since teratogenic potential of aprocitentan was investigated only at exposures slightly above the exposure at the maximum recommended human dose, it is not known which exposures may elicit adverse effects on embryo-foetal development.
In pre- and post-natal development studies, female rats treated from late pregnancy through lactation showed reduced pup survival and impairment of the reproductive capability of the offspring.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.