JEVTANA Concentrate and solvent for solution for infusion Ref.[6469] Active ingredients: Cabazitaxel
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Sanofi-aventis groupe, 54, rue La Boétie, F-75008 Paris, France
Contraindications
- Hypersensitivity to cabazitaxel, to other taxanes, or polysorbate 80 or any excipients listed in section 6.1.
- Neutrophil counts less than 1,500/mm³.
- Severe hepatic impairment (total bilirubin >3 x ULN).
- Concomitant vaccination with yellow fever vaccine (see section 4.5).
Special warnings and precautions for use
Hypersensitivity reactions
All patients should be pre-medicated prior to the initiation of the infusion of cabazitaxel (see section 4.2).
Patients should be observed closely for hypersensitivity reactions especially during the first and second infusions. Hypersensitivity reactions may occur within a few minutes following the initiation of the infusion of cabazitaxel, thus facilities and equipment for the treatment of hypotension and bronchospasm should be available. Severe reactions can occur and may include generalised rash/erythema, hypotension and bronchospasm. Severe hypersensitivity reactions require immediate discontinuation of cabazitaxel and appropriate therapy. Patients with a hypersensitivity reaction must stop treatment with JEVTANA (see section 4.3).
Bone marrow suppression
Bone marrow suppression manifested as neutropenia, anaemia, thrombocytopenia, or pancytopenia may occur (see “Risk of neutropenia” and “Anaemia” in section 4.4 below).
Risk of neutropenia
Patients treated with cabazitaxel may receive prophylactic G-CSF, as per American Society of Clinical Oncology (ASCO) guidelines and/or current institutional guidelines, to reduce the risk or manage neutropenia complications (febrile neutropenia, prolonged neutropenia or neutropenic infection). Primary prophylaxis with G-CSF should be considered in patients with high-risk clinical features (age >65 years, poor performance status, previous episodes of febrile neutropenia, extensive prior radiation ports, poor nutritional status, or other serious comorbidities) that predispose them to increased complications from prolonged neutropenia. The use of G-CSF has been shown to limit the incidence and severity of neutropenia.
Neutropenia is the most common adverse reaction of cabazitaxel (see section 4.8). Monitoring of complete blood counts is essential on a weekly basis during cycle 1 and before each treatment cycle thereafter so that the dose can be adjusted, if needed.
The dose should be reduced in case of febrile neutropenia, or prolonged neutropenia despite appropriate treatment (see section 4.2). Patients should be re-treated only when neutrophils recover to a level ≥1,500/mm³ (see section 4.3).
Gastrointestinal disorders
Symptoms such as abdominal pain and tenderness, fever, persistent constipation, diarrhoea, with or without neutropenia, may be early manifestations of serious gastrointestinal toxicity and should be evaluated and treated promptly. Cabazitaxel treatment delay or discontinuation may be necessary.
Risk of nausea, vomiting, diarrhoea and dehydration
If patients experience diarrhoea following administration of cabazitaxel they may be treated with commonly used anti-diarrhoeal medicinal products. Appropriate measures should be taken to re-hydrate patients. Diarrhoea can occur more frequently in patients that have received prior abdomino-pelvic radiation. Dehydration is more common in patients aged 65 or older. Appropriate measures should be taken to rehydrate patients and to monitor and correct serum electrolyte levels, particularly potassium. Treatment delay or dose reduction may be necessary for grade ≥3 diarrhoea (see section 4.2). If patients experience nausea or vomiting, they may be treated with commonly used anti-emetics.
Risk of serious gastrointestinal reactions
Gastrointestinal (GI) hemorrhage and perforation, ileus, colitis, including fatal outcome, have been reported in patients treated with cabazitaxel (see section 4.8). Caution is advised with treatment of patients most at risk of developing gastrointestinal complications: those with neutropenia, the elderly, concomitant use of NSAIDs, anti-platelet therapy or anti-coagulants, and patients with a prior history of pelvic radiotherapy or gastrointestinal disease, such as ulceration and GI bleeding.
Peripheral neuropathy
Cases of peripheral neuropathy, peripheral sensory neuropathy (e.g., paraesthesias, dysaesthesias) and peripheral motor neuropathy have been observed in patients receiving cabazitaxel. Patients under treatment with cabazitaxel should be advised to inform their doctor prior to continuing treatment if symptoms of neuropathy such as pain, burning, tingling, numbness, or weakness develop. Physicians should assess for the presence or worsening of neuropathy before each treatment. Treatment should be delayed until improvement of symptoms. The dose of cabazitaxel should be reduced from 25 mg/m² to 20 mg/m² for persistent grade >2 peripheral neuropathy (see section 4.2).
Anaemia
Anaemia has been observed in patients receiving cabazitaxel (see section 4.8). Haemoglobin and haematocrit should be checked before treatment with cabazitaxel and if patients exhibit signs or symptoms of anaemia or blood loss. Caution is recommended in patients with haemoglobin <10 g/dl and appropriate measures should be taken as clinically indicated.
Risk of renal failure
Renal disorders, have been reported in association with sepsis, severe dehydration due to diarrhoea, vomiting and obstructive uropathy. Renal failure including cases with fatal outcome has been observed. Appropriate measures should be taken to identify the cause and intensively treat the patients if this occurs.
Adequate hydration should be ensured throughout treatment with cabazitaxel. The patient should be advised to report any significant change in daily urinary volume immediately. Serum creatinine should be measured at baseline, with each blood count and whenever the patient reports a change in urinary output. Cabazitaxel treatment should be discontinued in case of any degradation of renal function to renal failure ≥CTCAE 4.0 Grade 3.
Respiratory disorders
Interstitial pneumonia/pneumonitis and interstitial lung disease have been reported and may be associated with fatal outcome (see section 4.8).
If new or worsening pulmonary symptoms develop, patients should be closely monitored, promptly investigated, and appropriately treated. Interruption of cabazitaxel therapy is recommended until diagnosis is available. Early use of supportive care measures may help improve the condition. The benefit of resuming cabazitaxel treatment must be carefully evaluated. Risk of cardiac arrhythmias Cardiac arrhythmias have been reported, most commonly tachycardia and atrial fibrillation (see section 4.8).
Elderly
Elderly people (≥65 years of age) may be more likely to experience certain adverse reactions including neutropenia and febrile neutropenia (see section 4.8).
Patients with liver impairment
Treatment with JEVTANA is contraindicated in patients with severe hepatic impairment (total bilirubin >3 x ULN) (See sections 4.3 and 5.2).
Dose should be reduced for patients with mild (total bilirubin >1 to ≤1.5 x ULN or AST >1.5 x ULN), hepatic impairment (see sections 4.2 and 5.2).
Interactions
Co-administration with strong CYP3A inhibitors should be avoided since they may increase the plasma concentrations of cabazitaxel (see sections 4.2 and 4.5). If co-administration with a strong CYP3A inhibitor cannot be avoided, close monitoring for toxicity and a cabazitaxel dose reduction should be considered (see sections 4.2 and 4.5).
Co-administration with strong CYP3A inducers should be avoided since they may decrease plasma concentrations of cabazitaxel (see sections 4.2 and 4.5).
Excipients
The solvent contains 573.3 mg ethanol 96% (15% v/v), equivalent to 14 ml of beer or 6 ml of wine. Harmful for those suffering from alcoholism.
To be taken into account in high-risk groups such as patients with liver disease, or epilepsy.
Interaction with other medicinal products and other forms of interaction
In vitro studies have shown that cabazitaxel is mainly metabolised through CYP3A (80% to 90%) (see section 5.2).
CYP3A inhibitors
Repeated administration of ketoconazole (400 mg once daily), a strong CYP3A inhibitor, resulted in a 20% decrease in cabazitaxel clearance corresponding to a 25% increase in AUC. Therefore concomitant administration of strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) should be avoided as an increase of plasma concentrations of cabazitaxel may occur (see sections 4.2 and 4.4).
Concomitant administration of aprepitant, a moderate CYP3A inhibitor, had no effect on cabazitaxel clearance.
CYP3A inducers
Repeated administration of rifampin (600 mg once daily), a strong CYP3A inducer, resulted in an increase in cabazitaxel clearance of 21% corresponding to a decrease in AUC of 17%. Therefore concomitant administration of strong CYP3A inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentin, phenobarbital) should be avoided as a decrease of plasma concentrations of cabazitaxel may occur (see sections 4.2 and 4.4). In addition, patients should also refrain from taking St. John’s Wort.
OATP1B1
In vitro, cabazitaxel has also been shown to inhibit the transport proteins of the Organic Anion Transport Polypeptides OATP1B1. The risk of interaction with OATP1B1 substrates (e.g. statins, valsartan, repaglinide) is possible, notably during the infusion duration (1 hour) and up to 20 minutes after the end of the infusion. A time interval of 12 hours is recommended before the infusion and at least 3 hours after the end of infusion before administering the OATP1B1 substrates.
Vaccinations
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents may result in serious or fatal infections. Vaccination with a live attenuated vaccine should be avoided in patients receiving cabazitaxel. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Pregnancy and lactation
Pregnancy
There are no data from the use of cabazitaxel in pregnant women. Studies in animals have shown reproductive toxicity at maternotoxic doses (see section 5.3) and that cabazitaxel crosses the placenta barrier (see section 5.3). As with other cytotoxic medicinal products, cabazitaxel may cause foetal harm in exposed pregnant women. Cabazitaxel is not recommended during pregnancy and in women of childbearing potential not using contraception.
Breast-feeding
Available pharmacokinetics data in animals have shown excretion of cabazitaxel and its metabolites in milk (see section 5.3). A risk to the suckling child cannot be excluded. Cabazitaxel should not be used during breast-feeding.
Fertility
Animal studies showed that cabazitaxel affected reproductive system in male rats and dogs without any functional effect on fertility (see section 5.3). Nevertheless, considering the pharmacological activity of taxanes, their genotoxic potential and effect of several compounds of this class on fertility in animal studies, effect on male fertility could not be excluded in human.
Due to potential effects on male gametes and to potential exposure via seminal liquid, men treated with cabazitaxel should use effective contraception throughout treatment and are recommended to continue this for up to 6 months after the last dose of cabazitaxel. Due to potential exposure via seminal liquid, men treated with cabazitaxel should prevent contact with the ejaculate by another person throughout treatment. Men being treated with cabazitaxel are advised to seek advice on conservation of sperm prior to treatment.
Effects on ability to drive and use machines
Cabazitaxel may influence the ability to drive and use machines as it may cause fatigue and dizziness. Patients should be advised not to drive or use machines if they experience these adverse reactions during treatment.
Undesirable effects
Summary of safety profile
The safety of JEVTANA in combination with prednisone or prednisolone was evaluated in 371 patients with metastatic castration resistant prostate cancer who were treated with 25 mg/m² cabazitaxel once every three weeks in a randomised open label, controlled phase III study. Patients received a median duration of 6 cycles of cabazitaxel.
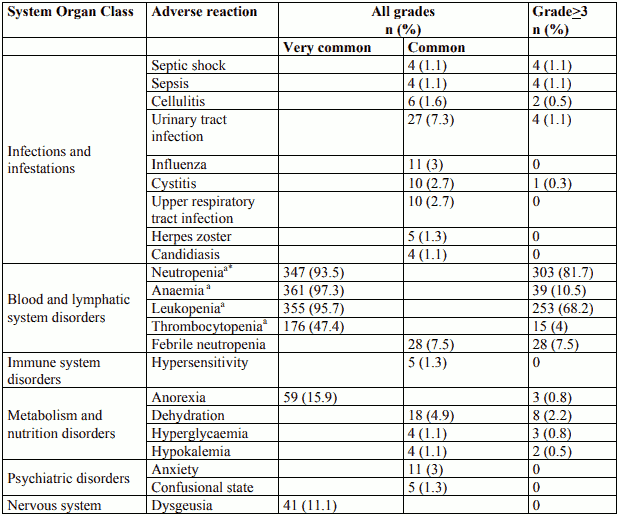
The most commonly (≥10%) occurring adverse reactions in all grades were anaemia (97.3%), leukopenia (95.7%), neutropenia (93.5%), thrombocytopenia (47.4%), and diarrhoea (46.6%). The most commonly (≥5%) occurring grade ≥3 adverse reactions in the cabazitaxel group were neutropenia (81.7%), leukopenia (68.2%), anaemia (10.5%), febrile neutropenia (7.5%), diarrhoea (6.2%).
Discontinuation of treatment due to adverse reactions occurred in 68 patients (18.3%) receiving cabazitaxel. The most common adverse reactions leading to cabazitaxel discontinuation was neutropenia.
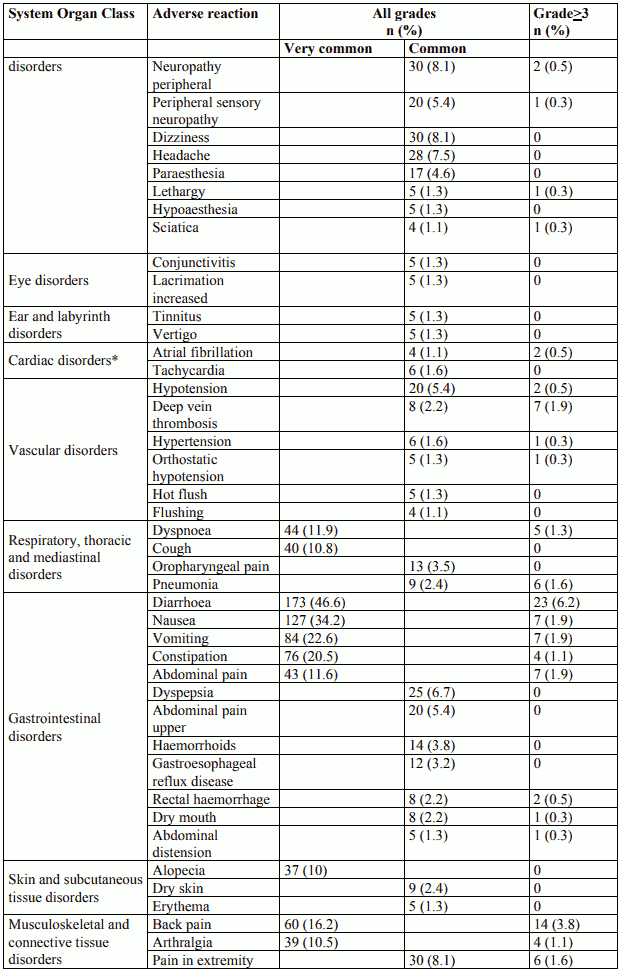
Tabulated list of adverse reactions
Adverse reactions are listed in table 2 according to MedDRA system organ class and frequency categories. Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. Intensity of the adverse reactions is graded according to CTCAE 4.0 (grade ≥3 = G≥3). Frequencies are based on all grades and defined as: very common (≥1/10), common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data).
Table 2. Reported adverse reactions and haematological abnormalities with cabazitaxel in combination with prednisone or prednisolone in the TROPIC study (n=371):

Description of selected adverse reactions
Neutropenia, and associated clinical events
Incidence of grade ≥3 neutropenia based on laboratory data was 81.7%. The incidence of grade ≥3 clinical neutropenia and febrile neutropenia adverse reactions were 21.3% and 7.5% respectively. Neutropenia was the most common adverse reaction leading to medicinal product discontinuation (2.4%).
Neutropenic complications included neutropenic infections (0.5%), neutropenic sepsis (0.8%), and septic shock (1.1%), which in some cases resulted in a fatal outcome. The use of G-CSF has been shown to limit the incidence and severity of neutropenia (see sections 4.2 and 4.4).
Cardiac disorders and arrhythmias
All Grade events among cardiac disorders were more common on cabazitaxel of which 6 patients (1.6%) had Grade ≥3 cardiac arrhythmias. The incidence of tachycardia on cabazitaxel was 1.6%, none of which were Grade ≥3. The incidence of atrial fibrillation was 1.1% in the cabazitaxel group. Cardiac failure events were more common on cabazitaxel, the event term being reported for 2 patients (0.5%). One patient in the cabazitaxel group died from cardiac failure. Fatal ventricular fibrillation was reported in 1 patient (0.3%), and cardiac arrest in 2 patients (0.5%). None were considered related by the investigator.
Haematuria
Haematuria all grades frequency was 20.8% at 25 mg/m² in EFC11785 study (see section 5.1). Confounding causes such as disease progression, instrumentation, infection or anticoagulation/NSAID/aspirin therapy were identified in nearly two thirds of the cases.
Other laboratory abnormalities
The incidence of grade ≥3 anaemia, increased AST, ALT, and bilirubin based on laboratory abnormalities were 10.5%, 0.7%, 0.9%, and 0.6%, respectively.
Gastrointestinal disorders
Colitis, enterocolitis, gastritis, neutropenic enterocolitis have been observed. Gastrointestinal hemorrhage and perforation, ileus and intestinal obstruction have also been reported (see section 4.4).
Respiratory disorders
Cases of interstitial pneumonia/pneumonitis and interstitial lung disease, sometimes fatal have been reported with an unknown frequency (cannot be estimated from the available data) (see section 4.4).
Renal and urinary disorders
Cystitis due to radiation recall phenomenon, including haemorrhagic cystitis, were reported uncommonly.
Paediatric population
See section 4.2
Other special populations
Elderly population
Among the 371 patients treated with cabazitaxel in the prostate cancer study, 240 patients were 65 years or over including 70 patients older than 75 years. The following adverse reactions reported at rates 5% higher in patients 65 years of age or greater compared to younger patients were fatigue (40.4% versus 29.8%), clinical neutropenia (24.2% versus 17.6%), asthenia (23.8% versus 14.5%), pyrexia (14.6% versus 7.6%), dizziness (10.0% versus 4.6%), urinary tract infection (9.6% versus 3.1%) and dehydration (6.7% versus 1.5%), respectively. The incidence of the following grade ≥3 adverse reactions were higher in patients 65 years of age compared to younger patients; neutropenia based on laboratory abnormalities (86.3% versus 73.3%), clinical neutropenia (23.8% versus 16.8%) and febrile neutropenia (8.3% versus 6.1%) (see sections 4.2 and 4.4).
Of the 595 patients treated with cabazitaxel 25 mg/m² in the prostate cancer EFC 11785 study, 420 patients were 65 years or over. The adverse reactions reported at rates of at least 5% higher in patients 65 years of age or greater compared to younger patients were diarrhoea (42.9% vs. 32.6%), fatigue (30.2% vs. 19.4%), asthenia (22.4% vs. 13.1%), constipation (20.2% vs. 12.6%), clinical neutropenia (12.9% vs. 6.3%), febrile neutropenia (11.2% vs. 4.6%) and dyspnoea (9.5% vs. 3.4%).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6.
PVC infusion containers or polyurethane infusion sets should not be used for the preparation and administration of the infusion solution.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.