KALETRA Film-coated tablet Ref.[107476] Active ingredients: Lopinavir Lopinavir and Ritonavir Ritonavir
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061 Ludwigshafen, Germany
4.3. Contraindications
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1.
Severe hepatic insufficiency.
Kaletra contains lopinavir and ritonavir, both of which are inhibitors of the P450 isoform CYP3A. Kaletra should not be co-administered with medicinal products that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life threatening events. These medicinal products include:
| Medicinal product class | Medicinal products within class | Rationale |
|---|---|---|
| Concomitant medicinal product levels increased | ||
| Alpha1-adrenoreceptor antagonist | Alfuzosin | Increased plasma concentrations of alfuzosin which may lead to severe hypotension. The concomitant administration with alfuzosin is contraindicated (see section 4.5). |
| Antianginal | Ranolazine | Increased plasma concentrations of ranolazine which may increase the potential for serious and/or life- threatening reactions (see section 4.5). |
| Antiarrhythmics | Amiodarone, dronedarone | Increased plasma concentrations of amiodarone and dronedarone. Thereby, increasing the risk of arrhythmias or other serious adverse reactions (see section 4.5). |
| Antibiotic | Fusidic Acid | Increased plasma concentrations of fusidic acid. The concomitant administration with fusidic acid is contraindicated in dermatological infections (see section 4.5). |
| Anticancer | Neratinib | Increased plasma concentrations of neratinib which may increase the potential for serious and/or life-threatening reactions (see section 4.5). |
| Venetoclax | Increased plasma concentrations of venetoclax. Increased risk of tumor lysis syndrome at the dose initiation and during the ramp-up phase (see section 4.5). | |
| Anti-gout | Colchicine | Increased plasma concentrations of colchicine. Potential for serious and/or life-threatening reactions in patients with renal and/or hepatic impairment (see sections 4.4 and 4.5). |
| Antihistamines | Astemizole, terfenadine | Increased plasma concentrations of astemizole and terfenadine. Thereby, increasing the risk of serious arrhythmias from these agents (see section 4.5). |
| Antipsychotics/ Neuroleptics | Lurasidone | Increased plasma concentrations of lurasidone which may increase the potential for serious and/or life- threatening reactions (see section 4.5). |
| Pimozide | Increased plasma concentrations of pimozide. Thereby, increasing the risk of serious haematologic abnormalities, or other serious adverse effects from this agent (see section 4.5). | |
| Quetiapine | Increased plasma concentrations of quetiapine which may lead to coma. The concomitant administration with quetiapine is contraindicated (see section 4.5). | |
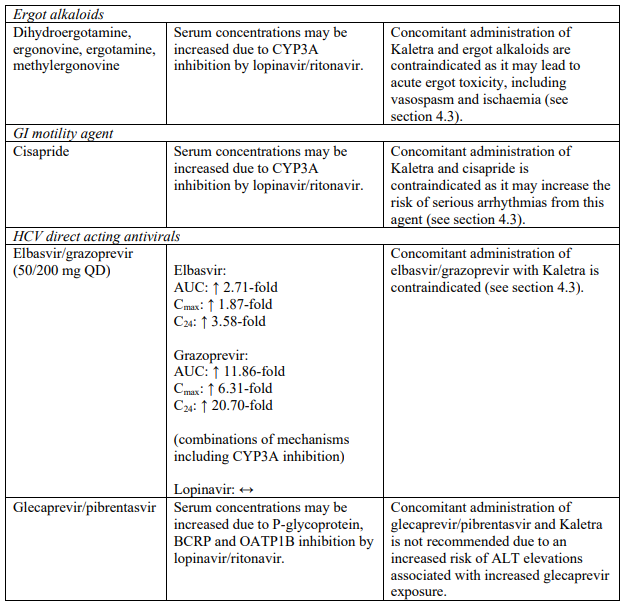
| Ergot alkaloids | Dihydroergotamine, ergonovine, ergotamine, methylergonovine | Increased plasma concentrations of ergot derivatives leading to acute ergot toxicity, including vasospasm and ischaemia (see section 4.5). |
| GI motility agent | Cisapride | Increased plasma concentrations of cisapride. Thereby, increasing the risk of serious arrhythmias from this agent (see section 4.5). |
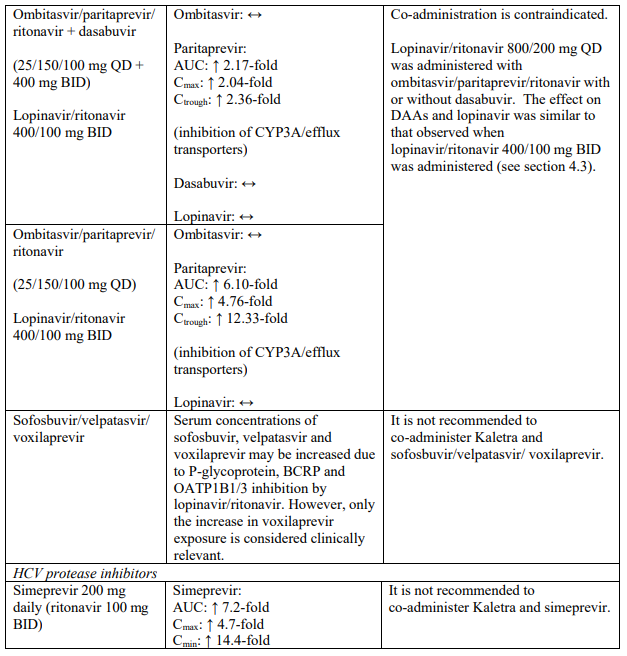
| Hepatitis C virus direct acting antivirals | Elbasvir/grazoprevir | Increased risk of alanine transaminase (ALT) elevations (see section 4.5). |
| Ombitasvir/paritaprevir/ritonavir with or without dasabuvir | Increased plasma concentrations of paritaprevir; thereby, increasing the risk of alanine transaminase (ALT) elevations (see section 4.5). | |
| Lipid-modifying agents | ||
| HMG Co-A Reductase Inhibitors | Lovastatin, simvastatin | Increased plasma concentrations of lovastatin and simvastatin; thereby, increasing the risk of myopathy including rhabdomyolysis (see section 4.5). |
| Microsomal triglyceride transfer protein (MTTP) inhibitor | Lomitapide | Increased plasma concentrations of lomitapide (see section 4.5). |
| Phosphodiesterase (PDE5) inhibitors | Avanafil | Increased plasma concentrations of avanafil (see sections 4.4 and 4.5). |
| Sildenafil | Contraindicated when used for the treatment of pulmonary arterial hypertension (PAH) only. Increased plasma concentrations of sildenafil. Thereby, increasing the potential for sildenafil-associated adverse events (which include hypotension and syncope). See section 4.4 and section 4.5 for co-administration of sildenafil in patients with erectile dysfunction. | |
| Vardenafil | Increased plasma concentrations of vardenafil (see sections 4.4 and 4.5) | |
| Sedatives/hypnotics | Oral midazolam, triazolam | Increased plasma concentrations of oral midazolam and triazolam. Thereby, increasing the risk of extreme sedation and respiratory depression from these agents. For caution on parenterally administered midazolam, see section 4.5. |
| Lopinavir/ritonavir medicinal product level decreased | ||
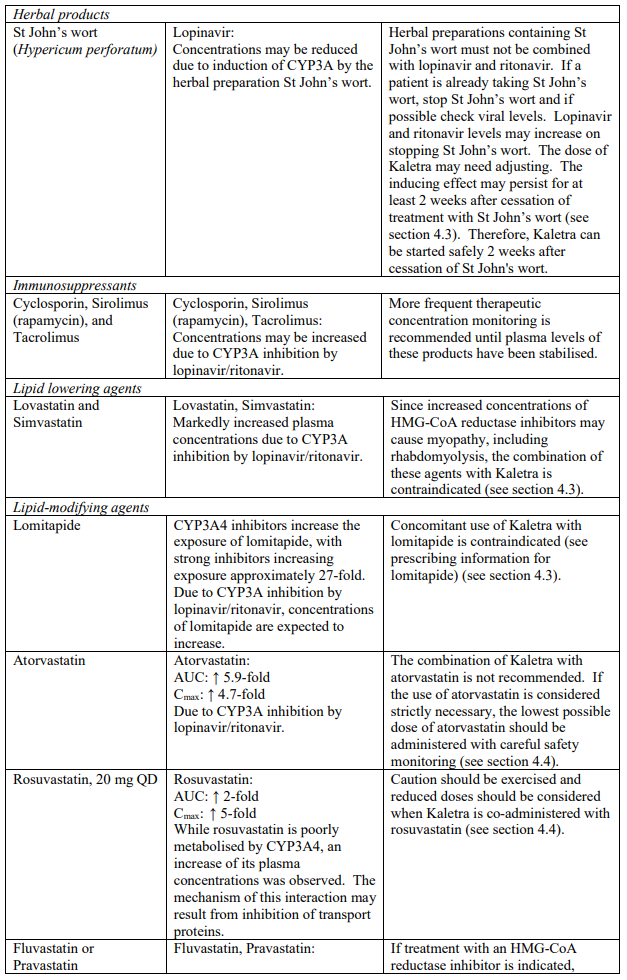
| Herbal products | St. John’s wort | Herbal preparations containing St John’s wort (Hypericum perforatum) due to the risk of decreased plasma concentrations and reduced clinical effects of lopinavir and ritonavir (see section 4.5). |
Kaletra oral solution is contraindicated in children below the age of 14 days, pregnant women, patients with hepatic or renal failure and patients treated with disulfiram or metronidazole due to the potential risk of toxicity from the excipient propylene glycol (see section 4.4).
4.4. Special warnings and precautions for use
Patients with coexisting conditions
Hepatic impairment
The safety and efficacy of Kaletra has not been established in patients with significant underlying liver disorders. Kaletra is contraindicated in patients with severe liver impairment (see section 4.3). Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reactions. In case of concomitant antiviral therapy for hepatitis B or C, please refer to the relevant product information for these medicinal products.
Patients with pre-existing liver dysfunction including chronic hepatitis have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment should be considered.
Elevated transaminases with or without elevated bilirubin levels have been reported in HIV-1 mono-infected and in individuals treated for post-exposure prophylaxis as early as 7 days after the initiation of lopinavir/ritonavir in conjunction with other antiretroviral agents. In some cases the hepatic dysfunction was serious.
Appropriate laboratory testing should be conducted prior to initiating therapy with lopinavir/ritonavir and close monitoring should be performed during treatment.
Renal impairment
Since the renal clearance of lopinavir and ritonavir is negligible, increased plasma concentrations are not expected in patients with renal impairment. Because lopinavir and ritonavir are highly protein bound, it is unlikely that they will be significantly removed by haemodialysis or peritoneal dialysis.
Haemophilia
There have been reports of increased bleeding, including spontaneous skin haematomas and haemarthrosis in patients with haemophilia type A and B treated with protease inhibitors. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced if treatment had been discontinued. A causal relationship had been evoked, although the mechanism of action had not been elucidated. Haemophiliac patients should therefore be made aware of the possibility of increased bleeding.
Pancreatitis
Cases of pancreatitis have been reported in patients receiving Kaletra, including those who developed hypertriglyceridaemia. In most of these cases patients have had a prior history of pancreatitis and/or concurrent therapy with other medicinal products associated with pancreatitis. Marked triglyceride elevation is a risk factor for development of pancreatitis. Patients with advanced HIV disease may be at risk of elevated triglycerides and pancreatitis.
Pancreatitis should be considered if clinical symptoms (nausea, vomiting, abdominal pain) or abnormalities in laboratory values (such as increased serum lipase or amylase values) suggestive of pancreatitis should occur. Patients who exhibit these signs or symptoms should be evaluated and Kaletra therapy should be suspended if a diagnosis of pancreatitis is made (see section 4.8).
Immune Reconstitution Inflammatory Syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymtomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis jiroveci pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the reported time to onset is more variable and can occur many months after initiation of treatment.
Osteonecrosis
Although the etiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
PR interval prolongation
Lopinavir/ritonavir has been shown to cause modest asymptomatic prolongation of the PR interval in some healthy adult subjects. Rare reports of 2nd or 3rd degree atroventricular block in patients with underlying structural heart disease and pre-existing conduction system abnormalities or in patients receiving drugs known to prolong the PR interval (such as verapamil or atazanavir) have been reported in patients receiving lopinavir/ritonavir. Kaletra should be used with caution in such patients (see section 5.1).
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose, reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Interactions with medicinal products
Kaletra contains lopinavir and ritonavir, both of which are inhibitors of the P450 isoform CYP3A. Kaletra is likely to increase plasma concentrations of medicinal products that are primarily metabolised by CYP3A. These increases of plasma concentrations of co-administered medicinal products could increase or prolong their therapeutic effect and adverse events (see sections 4.3 and 4.5).
Strong CYP3A4 inhibitors such as protease inhibitors may increase bedaquiline exposure which could potentially increase the risk of bedaquiline-related adverse reactions. Therefore, combination of bedaquiline with lopinavir/ritonavir should be avoided. However, if the benefit outweighs the risk, co-administration of bedaquiline with lopinavir/ritonavir must be done with caution. More frequent electrocardiogram monitoring and monitoring of transaminases is recommended (see section 4.5 and refer to the bedaquiline SmPC).
Co-administration of delamanid with a strong inhibitor of CYP3A (as lopinavir/ritonavir) may increase exposure to delamanid metabolite, which has been associated with QTc prolongation. Therefore, if co-administration of delamanid with lopinavir/ritonavir is considered necessary, very frequent ECG monitoring throughout the full delamanid treatment period is recommended (see section 4.5 and refer to the delamanid SmPC).
Life-threatening and fatal drug interactions have been reported in patients treated with colchicine and strong inhibitors of CYP3A like ritonavir. Concomitant administration with colchicine is contraindicated in patients with renal and/or hepatic impairment (see sections 4.3 and 4.5).
The combination of Kaletra with:
- tadalafil, indicated for the treatment of pulmonary arterial hypertension, is not recommended (see section 4.5);
- riociguat is not recommended (see section 4.5);
- vorapaxar is not recommended (see section 4.5);
- fusidic acid in osteo-articular infections is not recommended (see section 4.5);
- salmeterol is not recommended (see section 4.5);
- rivaroxaban is not recommended (see section 4.5).
The combination of Kaletra with atorvastatin is not recommended. If the use of atorvastatin is considered strictly necessary, the lowest possible dose of atorvastatin should be administered with careful safety monitoring. Caution must also be exercised and reduced doses should be considered if Kaletra is used concurrently with rosuvastatin. If treatment with a HMG-CoA reductase inhibitor is indicated, pravastatin or fluvastatin is recommended (see section 4.5).
PDE5 inhibitors
Particular caution should be used when prescribing sildenafil or tadalafil for the treatment of erectile dysfunction in patients receiving Kaletra. Co-administration of Kaletra with these medicinal products is expected to substantially increase their concentrations and may result in associated adverse events such as hypotension, syncope, visual changes and prolonged erection (see section 4.5). Concomitant use of avanafil or vardenafil and lopinavir/ritonavir is contraindicated (see section 4.3). Concomitant use of sildenafil prescribed for the treatment of pulmonary arterial hypertension with Kaletra is contraindicated (see section 4.3).
Particular caution must be used when prescribing Kaletra and medicinal products known to induce QT interval prolongation such as: chlorpheniramine, quinidine, erythromycin, clarithromycin. Indeed, Kaletra could increase concentrations of the co-administered medicinal products and this may result in an increase of their associated cardiac adverse reactions. Cardiac events have been reported with Kaletra in preclinical studies; therefore, the potential cardiac effects of Kaletra cannot be currently ruled out (see sections 4.8 and 5.3).
Co-administration of Kaletra with rifampicin is not recommended. Rifampicin in combination with Kaletra causes large decreases in lopinavir concentrations which may in turn significantly decrease the lopinavir therapeutic effect. Adequate exposure to lopinavir/ritonavir may be achieved when a higher dose of Kaletra is used but this is associated with a higher risk of liver and gastrointestinal toxicity. Therefore, this co-administration should be avoided unless judged strictly necessary (see section 4.5).
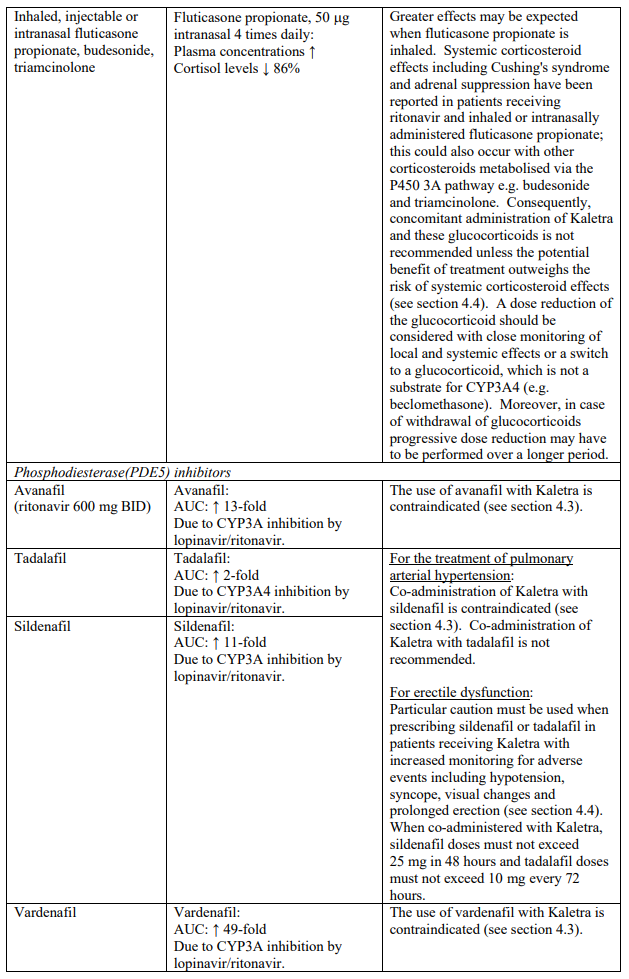
Concomitant use of Kaletra and fluticasone or other glucocorticoids that are metabolised by CYP3A4, such as budesonide and triamcinolone, is not recommended unless the potential benefit of treatment outweighs the risk of systemic corticosteroid effects, including Cushing’s syndrome and adrenal suppression (see section 4.5).
Other
Kaletra is not a cure for HIV infection or AIDS. People taking Kaletra may still develop infections or other illnesses associated with HIV disease and AIDS.
Sodium
This medicine contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially ‘sodiumfree’.
4.5. Interaction with other medicinal products and other forms of interaction
Kaletra contains lopinavir and ritonavir, both of which are inhibitors of the P450 isoform CYP3A in vitro. Co-administration of Kaletra and medicinal products primarily metabolised by CYP3A may result in increased plasma concentrations of the other medicinal product, which could increase or prolong its therapeutic and adverse reactions. Kaletra does not inhibit CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 or CYP1A2 at clinically relevant concentrations (see section 4.3).
Kaletra has been shown in vivo to induce its own metabolism and to increase the biotransformation of some medicinal products metabolised by cytochrome P450 enzymes (including CYP2C9 and CYP2C19) and by glucuronidation. This may result in lowered plasma concentrations and potential decrease of efficacy of co-administered medicinal products.
Medicinal products that are contraindicated specifically due to the expected magnitude of interaction and potential for serious adverse events are listed in section 4.3.
All interaction studies, when otherwise not stated, were performed using Kaletra capsules, which gives an approximately 20% lower exposure of lopinavir than the 200/50 mg tablets.
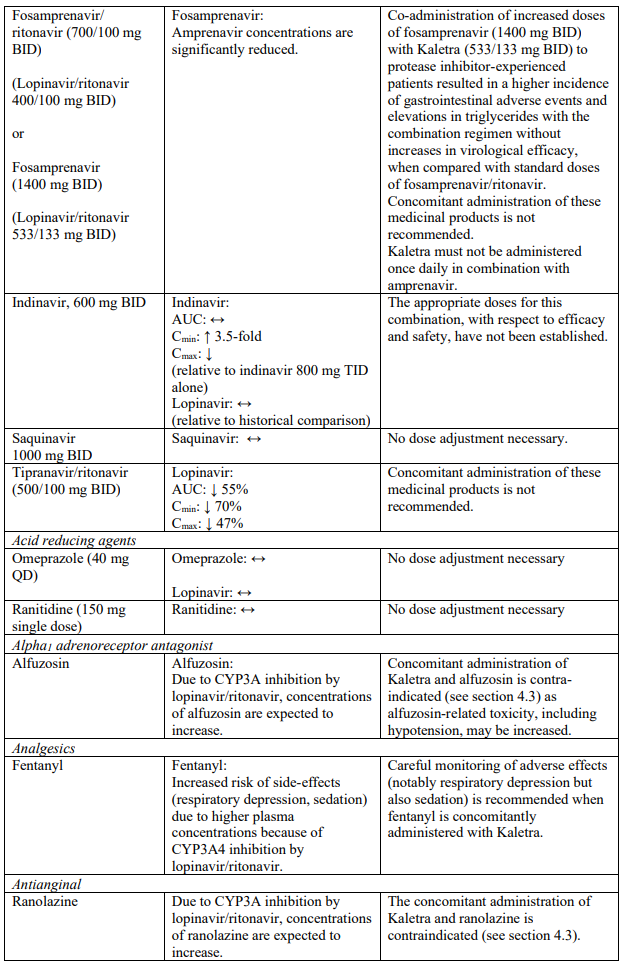
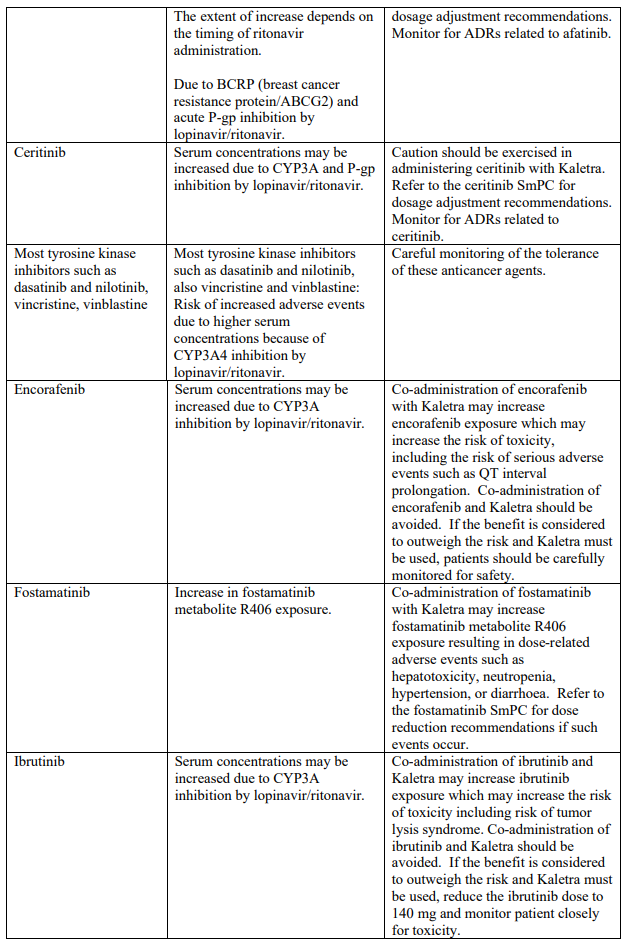
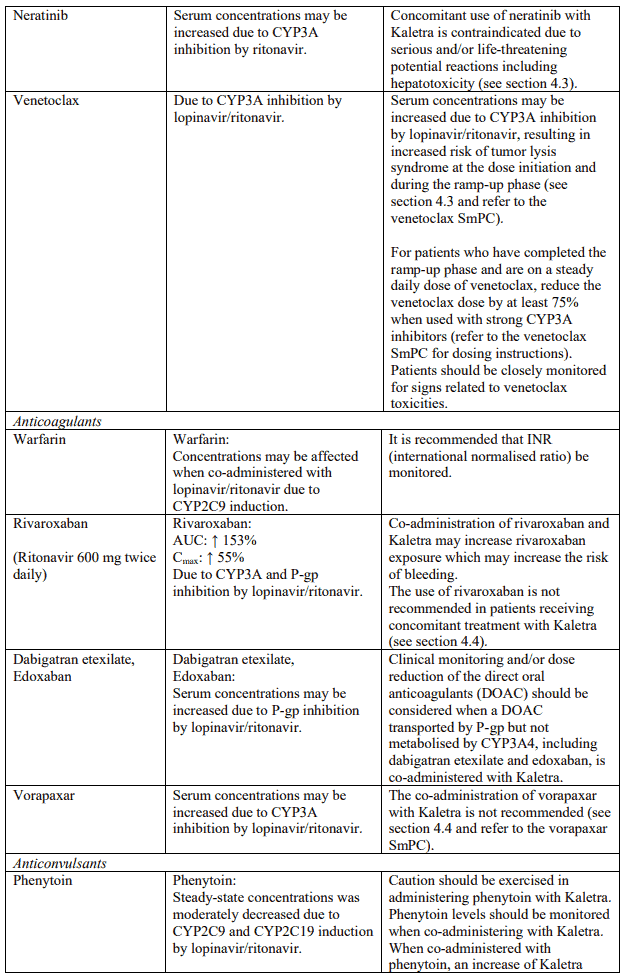
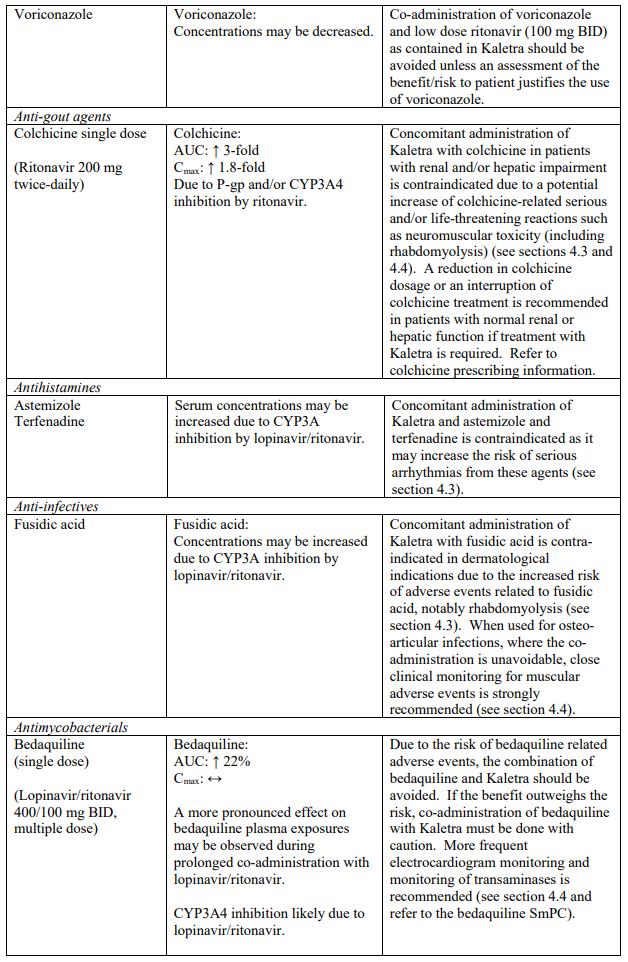
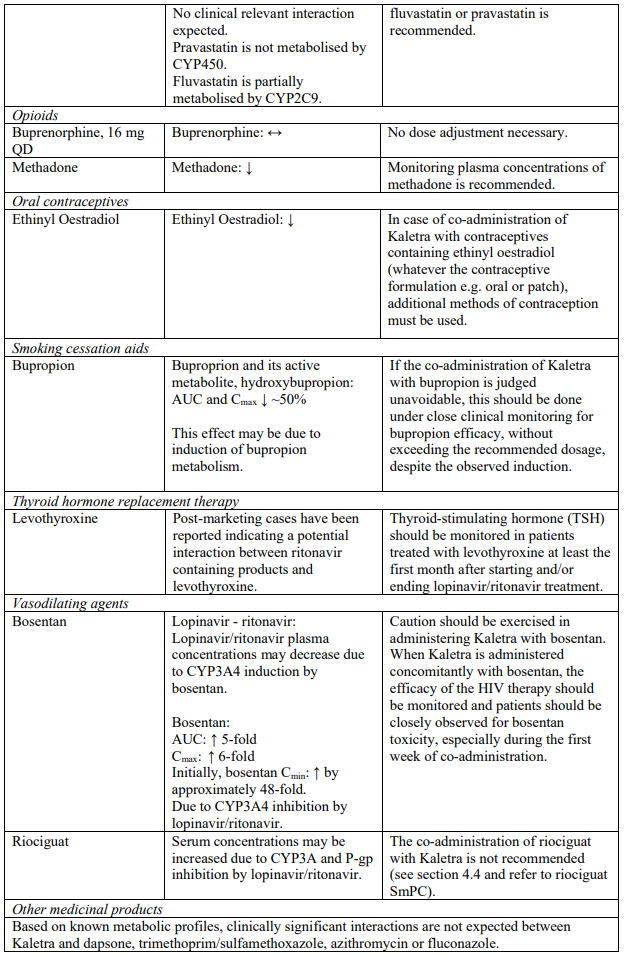
Known and theoretical interactions with selected antiretrovirals and non-antiretroviral medicinal products are listed in the table below. This list is not intended to be inclusive or comprehensive. Individual SmPCs should be consulted.
Interaction table
Interactions between Kaletra and co-administered medicinal products are listed in the table below (increase is indicated as “↑”, decrease as “↓”, no change as “↔”, once daily as “QD”, twice daily as “BID” and three times daily as “TID”).
Unless otherwise stated, studies detailed below have been performed with the recommended dosage of lopinavir/ritonavir (i.e. 400/100 mg twice daily).






4.6. Fertility, pregnancy and lactation
Pregnancy
As a general rule, when deciding to use antiretroviral agents for the treatment of HIV infection in pregnant women and consequently for reducing the risk of HIV vertical transmission to the newborn, the animal data as well as the clinical experience in pregnant women should be taken into account in order to characterise the safety for the foetus.
Lopinavir/ritonavir has been evaluated in over 3000 women during pregnancy, including over 1000 during the first trimester.
In post-marketing surveillance through the Antiretroviral Pregnancy Registry, established since January 1989, an increased risk of birth defects exposures with Kaletra has not been reported among over 1000 women exposed during the first trimester. The prevalence of birth defects after any trimester exposure to lopinavir is comparable to the prevalence observed in the general population. No pattern of birth defects suggestive of a common etiology was seen. Studies in animals have shown reproductive toxicity (see section 5.3). Based on the data mentioned, the malformative risk is unlikely in humans. Lopinavir can be used during pregnancy if clinically needed.
Breast-feeding
Studies in rats revealed that lopinavir is excreted in the milk. It is not known whether this medicinal product is excreted in human milk. As a general rule, it is recommended that women living with HIV do not breast-feed their babies in order to avoid transmission of HIV.
Fertility
Animal studies have shown no effects on fertility. No human data on the effect of lopinavir/ritonavir on fertility are available.
4.7. Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. Patients should be informed that nausea has been reported during treatment with Kaletra (see section 4.8).
4.8. Undesirable effects
a. Summary of the safety profile
The safety of Kaletra has been investigated in over 2600 patients in Phase II-IV clinical trials, of which over 700 have received a dose of 800/200 mg (6 capsules or 4 tablets) once daily. Along with nucleoside reverse transcriptase inhibitors (NRTIs), in some studies, Kaletra was used in combination with efavirenz or nevirapine.
The most common adverse reactions related to Kaletra therapy during clinical trials were diarrhoea, nausea, vomiting, hypertriglyceridaemia and hypercholesterolemia. Diarrhoea, nausea and vomiting may occur at the beginning of the treatment while hypertriglyceridaemia and hypercholesterolemia may occur later. Treatment emergent adverse events led to premature study discontinuation for 7% of subjects from Phase II-IV studies.
It is important to note that cases of pancreatitis have been reported in patients receiving Kaletra, including those who developed hypertriglyceridaemia. Furthermore, rare increases in PR interval have been reported during Kaletra therapy (see section 4.4).
b. Tabulated list of adverse reactions
Adverse reactions from clinical trials and post-marketing experience in adult and paediatric patients
The following events have been identified as adverse reactions. The frequency category includes all reported events of moderate to severe intensity, regardless of the individual causality assessment. The adverse reactions are displayed by system organ class. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1000 to <1/100), rare (≥1/10,000 to <1/1000) and not known (cannot be estimated from the available data).
Undesirable effects in clinical studies and post-marketing in adult patients:
| System organ class | Frequency | Adverse reaction |
|---|---|---|
| Infections and infestations | Very common | Upper respiratory tract infection |
| Common | Lower respiratory tract infection, skin infections including cellulitis, folliculitis and furuncle | |
| Blood and lymphatic system disorders | Common | Anaemia, leucopenia, neutropenia, lymphadenopathy |
| Immune system disorders | Common | Hypersensitivity including urticaria and angioedema |
| Uncommon | Immune reconstitution inflammatory syndrome | |
| Endocrine disorders | Uncommon | Hypogonadism |
| Metabolism and nutrition disorders | Common | Blood glucose disorders including diabetes mellitus, hypertriglyceridaemia, hypercholesterolemia, weight decreased, decreased appetite |
| Uncommon | Weight increased, increased appetite | |
| Psychiatric disorders | Common | Anxiety |
| Uncommon | Abnormal dreams, libido decreased | |
| Nervous system disorders | Common | Headache (including migraine), neuropathy (including peripheral neuropathy), dizziness, insomnia |
| Uncommon | Cerebrovascular accident, convulsion, dysgeusia, ageusia, tremor\ | |
| Eye disorders | Uncommon | Visual impairment |
| Ear and labyrinth disorders | Uncommon | Tinnitus, vertigo |
| Cardiac disorders | Uncommon | Atherosclerosis such as myocardial infarction1, atrioventricular block, tricuspid valve incompetence |
| Vascular disorders | Common | Hypertension |
| Uncommon | Deep vein thrombosis | |
| Gastrointestinal disorders | Very common | Diarrhoea, nausea |
| Common | Pancreatitis1, vomiting, gastrooesophageal reflux disease, gastroenteritis and colitis, abdominal pain (upper and lower), abdominal distension, dyspepsia, haemorrhoids, flatulence | |
| Uncommon | Gastrointestinal haemorrhage including gastrointestinal ulcer, duodenitis, gastritis and rectal haemorrhage, stomatitis and oral ulcers, faecal incontinence, constipation, dry mouth | |
| Hepatobiliary disorders | Common | Hepatitis including AST, ALT and GGT increases |
| Uncommon | Jaundice, hepatic steatosis, hepatomegaly, cholangitis, hyperbilirubinemia | |
| Skin and subcutaneous tissue disorders | Common | Rash including maculopapular rash, dermatitis/rash including eczema and seborrheic dermatitis, night sweats, pruritus |
| Uncommon | Alopecia, capillaritis, vasculitis | |
| Rare | Steven-Johnson syndrome, erythema multiforme | |
| Musculoskeletal and connective tissue disorders | Common | Myalgia, musculoskeletal pain including arthralgia and back pain, muscle disorders such as weakness and spasms |
| Uncommon | Rhabdomyolysis, osteonecrosis | |
| Renal and urinary disorders | Uncommon | Creatinine clearance decreased, nephritis, haematuria |
| Not known | Nephrolithiasis | |
| Reproductive system and breast disorders | Common | Erectile dysfunction, menstrual disorders - amenorrhoea, menorrhagia |
| General disorders and administration site conditions | Common | Fatigue including asthenia |
1 See section 4.4: pancreatitis and lipids
c. Description of selected adverse reactions
Cushing’s syndrome has been reported in patients receiving ritonavir and inhaled or intranasally administered fluticasone propionate; this could also occur with other corticosteroids metabolised via the P450 3A pathway e.g. budesonide (see section 4.4 and 4.5).
Increased creatine phosphokinase (CPK), myalgia, myositis, and rarely, rhabdomyolysis have been reported with protease inhibitors, particularly in combination with nucleoside reverse transcriptase inhibitors.
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and can occur many months after initiation of treatment (see section 4.4).
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
d. Paediatric populations
In children 14 days of age and older, the nature of the safety profile is similar to that seen in adults (see Table in section b).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.