KERENDIA Film-coated tablet Ref.[49668] Active ingredients: Finerenone
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Bayer AG, 51368 Leverkusen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: diuretics, aldosterone antagonists
ATC code: C03DA05
Mechanism of action
Finerenone is a nonsteroidal, selective antagonist of the mineralocorticoid receptor (MR) which is activated by aldosterone and cortisol and regulates gene transcription. Its binding to the MR leads to a specific receptor-ligand complex that blocks recruitment of transcriptional coactivators implicated in the expression of pro-inflammatory and pro-fibrotic mediators.
Pharmacodynamic effects
In FIDELIO-DKD, a randomised, double-blind, placebo-controlled, multicentre phase III study in adult patients with CKD and T2D, the placebo-corrected relative reduction in urinary albumin-to-creatinine ratio (UACR) in patients randomised to finerenone was 31% at month 4.
In ARTS-DN, a randomised, double-blind, placebo-controlled, multicentre phase IIb study in adult patients with CKD and T2D, the placebo-corrected relative reduction in UACR at Day 90 was 25% and 38% in patients treated with finerenone 10 mg and 20 mg once daily, respectively.
Cardiac electrophysiology
A dedicated QT study in 57 healthy participants showed that finerenone has no effect on cardiac repolarisation. There was no indication of a QT/QTc prolonging effect of finerenone after single doses of 20 mg (therapeutic) or 80 mg (supratherapeutic).
Clinical efficacy and safety
The FIDELIO-DKD study investigated the effect of finerenone compared to placebo on kidney and cardiovascular (CV) outcomes in adult patients with CKD and T2D. Patients were eligible based on evidence of persistent albuminuria (>30 mg/g to 5,000 mg/g), an eGFR of 25 to 75 mL/min/1.73 m², serum potassium ≤4.8 mmol/L at screening, and were required to be receiving standard of care, including a maximum tolerated labelled dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB). Patients with diagnosed heart failure with reduced ejection fraction and New York Heart Association II-IV were excluded due to the class 1A recommendation for MRA therapy.
The primary endpoint was a composite of time to first occurrence of kidney failure (defined as chronic dialysis or kidney transplantation, or a sustained decrease in eGFR to <15 mL/min/1.73 m² over at least 4 weeks), a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death. The key secondary endpoint was a composite of time to first occurrence of CV death, non-fatal myocardial infarction (MI), non-fatal stroke or hospitalisation for heart failure.
A total of 5,674 patients were randomised to receive either finerenone (N=2,833) or placebo (N=2,841) and included in the analyses. The median follow-up was 2.6 years. The dose of finerenone or placebo could be adjusted between 10 mg and 20 mg once daily during the course of the study, based mainly on serum potassium concentration. At month 24, of the subjects treated with finerenone, 67% were treated with 20 mg once daily, 30% with 10 mg once daily and 3% were on a treatment interruption.
After the end of study, vital status was obtained for 99.7% of patients. The study population was 63% White, 25% Asian and 5% Black. The mean age at enrolment was 66 years and 70% of patients were male. At baseline, the mean eGFR was 44.3 mL/min/1.73 m², with 55% of patients having an eGFR <45 mL/min/1.73 m², median UACR was 852 mg/g, and mean HbA1c was 7.7%, 46% had a history of atherosclerotic CV disease, 30% a history of coronary artery disease, 8% a history of cardiac failure, and the mean blood pressure was 138/76 mm Hg. The mean duration of T2D at baseline was 16.6 years and a history of diabetic retinopathy and diabetic neuropathy was reported in 47% and 26% of patients at baseline, respectively. At baseline, almost all patients were on ACEi (34%) or ARB (66%), and 97% of patients used one or more antidiabetic medications (insulin [64%], biguanides [44%], glucagon-like peptide-1 [GLP-1] receptor agonists [7%], sodium-glucose cotransporter 2 [SGLT2] inhibitors [5%]). The other most frequent medications taken at baseline were statins (74%) and calcium channel blockers (63%).
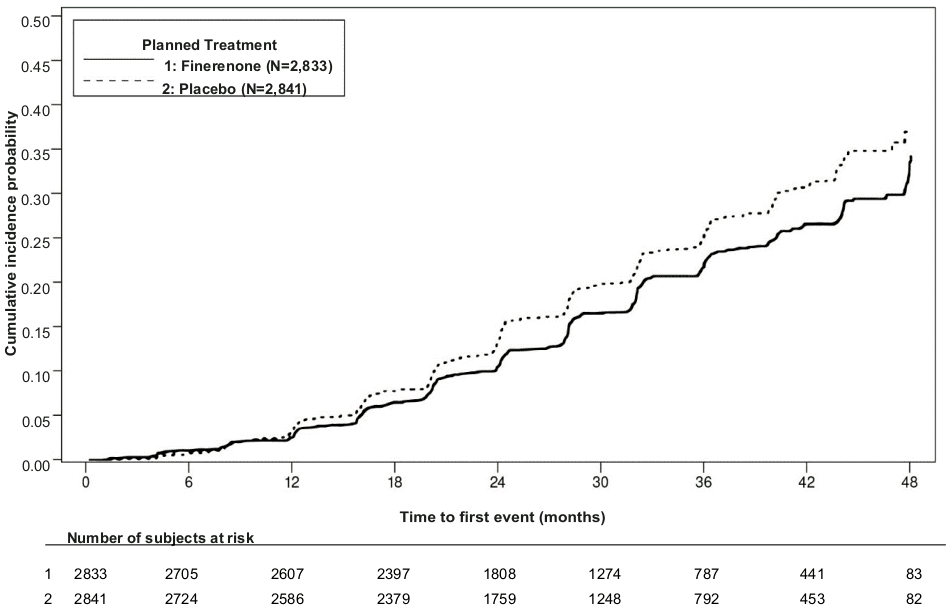
A statistically significant difference in favour of finerenone was shown for the primary composite endpoint and the key secondary composite endpoint (see figure 1/table 4 below). For the secondary endpoint of change in UACR from baseline to month 4, a relative reduction of 31.2% was observed in the finerenone group compared to placebo. The treatment effect for the primary and key secondary endpoints was generally consistent across subgroups, including region, eGFR, UACR, systolic blood pressure (BP) and HbA1c at baseline.
Table 4. Analysis of the primary and secondary time-to-event endpoints (and their individual components) in phase III study FIDELIO-DKD:
| Kerendia* (N=2,833) | Placebo (N=2,841) | Treatment effect | |||
|---|---|---|---|---|---|
| N (%) | Events/100-pyr | N (%) | Events/100-pyr | HR (95% CI) | |
| Primary renal composite endpoint and its components | |||||
| Composite of kidney failure, sustained eGFR decline ≥40% or renal death | 504 (17.8) | 7.59 | 600 (21.1) | 9.08 | 0.82 (0.73; 0.93) p=0.0014 |
| Kidney failure | 208 (7.3) | 2.99 | 235 (8.3) | 3.39 | 0.87 (0.72; 1.05) |
| Sustained eGFR decline 40% | 479 (16.9) | 7.21 | 577 (20.3) | 8.73 | 0.81 (0.72; 0.92) |
| Renal death | 2 (<0.1) | - | 2 (<0.1) | - | - |
| Key secondary CV composite endpoint and its components | |||||
| Composite of CV death, non-fatal MI, non-fatal stroke or hospitalisation for heart failure | 367 (13.0) | 5.11 | 420 (14.8) | 5.92 | 0.86 (0.75; 0.99) p=0.0339 |
| CV death | 128 (4.5) | 1.69 | 150 (5.3) | 1.99 | 0.86 (0.68;1.08) |
| Non-fatal MI | 70 (2.5) | 0.94 | 87 (3.1) | 1.17 | 0.80 (0.58;1.09) |
| Non-fatal stroke | 90 (3.2) | 1.21 | 87 (3.1) | 1.18 | 1.03 (0.76;1.38) |
| Hospitalisation for heart failure | 139 (4.9) | 1.89 | 162 (5.7) | 2.21 | 0.86 (0.68;1.08) |
| Secondary efficacy endpoints | |||||
| All-cause mortality | 219 (7.7) | 2.90 | 244 (8.6) | 3.23 | 0.90 (0.75; 1.07)** |
| All-cause hospitalisation | 1,263 (44.6) | 22.56 | 1,321 (46.5) | 23.87 | 0.95 (0.88; 1.02)** |
| Kidney failure, sustained eGFR decline ≥57% or renal death | 252 (8.9) | 3.64 | 326 (11.5) | 4.74 | 0.76 (0.65; 0.90)** |
* Treatment with 10 or 20 mg once daily in addition to maximum tolerated labelled doses of ACEi or ARB.
** p = not statistically significant after adjustment for multiplicity
CI: Confidence interval
HR: Hazard ratio
pyr: patient-years
Figure 1. Time to first occurrence of kidney failure, sustained decline in eGFR ≥40% from baseline, or renal death in the FIDELIO-DKD study:
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Kerendia in one or more subsets of the paediatric population in treatment of chronic kidney disease (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Finerenone is almost completely absorbed after oral administration. Absorption is rapid with maximum plasma concentrations (Cmax) appearing between 0.5 and 1.25 hours after tablet intake in the fasted state. The absolute bioavailability of finerenone is 43.5% due to first-pass metabolism in the gut-wall and liver. Finerenone is a substrate of the efflux transporter P-glycoprotein in vitro, which is however not considered relevant for its absorption in vivo due to the high permeability of finerenone.
Effect of food
Intake with high fat, high calorie food increased finerenone exposure AUC by 21%, reduced Cmax by 19% and prolonged the time to reach Cmax to 2.5 hours. Since this is not considered as clinically relevant, finerenone can be taken with or without food.
Distribution
The volume of distribution at steady state (Vss) of finerenone is 52.6 L. The human plasma protein binding of finerenone in vitro is 91.7%, with serum albumin being the main binding protein.
Biotransformation
Approximately 90% metabolism is mediated by CYP3A4 and 10% by CYP2C8. Four major metabolites were found in plasma. All metabolites are pharmacologically inactive.
Elimination
The elimination of finerenone from plasma is rapid with an elimination half-life (t½) of about 2 to 3 hours. Systemic blood clearance of finerenone is about 25 L/h. About 80% of the administered dose was excreted via urine and approximately 20% of the dose was excreted via faeces. Excretion was almost exclusively in the form of metabolites, while excretion of unchanged finerenone represents a minor route (<1% of dose in the urine due to glomerular filtration, <0.2% in the faeces).
Linearity
Finerenone pharmacokinetics are linear across the investigated dose range from 1.25 to 80 mg given as single dose tablets.
Special populations
Elderly
Of the 2,827 patients who received finerenone in the FIDELIO-DKD study, 58% of patients were 65 years and older, and 15% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
In a phase I study (N=48) elderly patients (≥65 years of age) exhibited higher finerenone plasma concentrations than younger patients (≤45 years of age), with mean AUC and Cmax values being 34% and 51% higher in the elderly (see section 4.2). Population-pharmacokinetic analyses did not identify age as a covariate for finerenone AUC or Cmax.
Renal impairment
Mild renal impairment (creatinine clearance [CLCR] 60 to <90 mL/min) did not affect finerenone AUC and Cmax. Compared to patients with normal renal function (CLCR ≥90 mL/min), the effect of moderate (CLCR 30 to <60 mL/min) or severe (CLCR <30 mL/min) renal impairment on AUC of finerenone was similar with increases by 34-36%. Moderate or severe renal impairment had no effect on Cmax (see section 4.2). Due to the high plasma protein binding, finerenone is not expected to be dialysable.
Hepatic impairment
There was no change in finerenone exposure in cirrhotic patients with mild hepatic impairment (see section 4.2). In cirrhotic patients with moderate hepatic impairment, finerenone total and unbound AUC were increased by 38% and 55%, respectively, while no change in Cmax was observed compared to healthy control participants (see section 4.2). There are no data in patients with severe hepatic impairment (see sections 4.2 and 4.5).
Body weight
Population-pharmacokinetic analyses identified body weight as a covariate for finerenone Cmax. The Cmax of a subject with a body weight of 50 kg was estimated to be 43% to 51% higher compared to a subject of 100 kg. Dose adaptation based on body weight is not warranted (see section 4.2).
Pharmacokinetic/pharmacodynamic relationships
The concentration-effect relationship over time for UACR was characterised by a maximum effect model indicating saturation at high exposures. The model-predicted time to reach the full (99%) steady-state drug effect on UACR was 138 days. The pharmacokinetic (PK) half-life was 2-3 hours and PK steady state was achieved after 2 days, indicating an indirect and delayed effect on pharmacodynamic responses.
Clinical studies with no relevant drug-drug interactions
Concomitant use of gemfibrozil (600 mg twice daily), a strong inhibitor of CYP2C8, increased finerenone mean AUC and Cmax 1.1-fold and 1.2-fold, respectively. This is not considered as clinically relevant.
Pre- and co-treatment with the proton pump inhibitor omeprazole (40 mg once daily) had no effect on finerenone mean AUC and mean Cmax. Concomitant use of antacid aluminium hydroxide and magnesium hydroxide (70 mVal) had no effect on finerenone mean AUC and reduced its mean Cmax by 19%. This is not considered as clinically relevant.
In vivo a multiple-dose regimen of 20 mg finerenone given once daily for 10 days had no relevant effect on the AUC of the CYP3A4 probe substrate midazolam. Therefore, a clinically relevant inhibition or induction of CYP3A4 by finerenone can be excluded.
A single dose of 20 mg finerenone also had no clinically relevant effect on AUC and Cmax of the CYP2C8 probe substrate repaglinide. Thus, finerenone does not inhibit CYP2C8.
Lack of mutual pharmacokinetic interaction was demonstrated between finerenone and the CYP2C9 substrate warfarin and between finerenone and the P-gp substrate digoxin.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, single dose toxicity, repeated dose toxicity, genotoxicity, phototoxicity, carcinogenic potential and male and female fertility.
Repeated dose toxicity
In dogs, a reduced prostate weight and size was found at an AUCunbound of about 10 to 60 times that in humans. The dose free of findings provides a safety margin of about 2.
Carcinogenic potential
In 2-year carcinogenicity studies, finerenone did not show carcinogenic potential in male and female rats or female mice. In male mice, finerenone resulted in an increase in Leydig cell adenoma at doses representing 26 times the AUCunbound in humans. A dose representing 17 times the AUCunbound in humans did not cause any tumours. Based on the known sensitivity of rodents to develop these tumours and the pharmacology-based mechanism at supratherapeutic doses as well as adequate safety margins, the increase in Leydig cell tumours in male mice is not clinically relevant.
Toxicity to development
In the embryo-foetal toxicity study in rats, finerenone resulted in reduced placental weights and signs of foetal toxicity, including reduced foetal weights and retarded ossification at the maternal toxic dose of 10 mg/kg/day corresponding to an AUCunbound of 19 times that in humans. At 30 mg/kg/day, the incidence of visceral and skeletal variations was increased (slight oedema, shortened umbilical cord, slightly enlarged fontanelle) and one foetus showed complex malformations including a rare malformation (double aortic arch) at an AUCunbound of about 25 times that in humans. The doses free of any findings (low dose in rats, high dose in rabbits) provided safety margins of 10 to 13 times for AUCunbound. Therefore, the findings in rats do not indicate an increased concern for foetal harm.
When rats were exposed during pregnancy and lactation in the pre- and postnatal developmental toxicity study, increased pup mortality and other adverse effects (lower pup weight, delayed pinna unfolding) were observed at about 4 times the AUCunbound expected in humans. In addition, the offspring showed slightly increased locomotor activity, but no other neurobehavioural changes starting at about 4 times the AUCunbound expected in humans. The dose free of findings provided a safety margin of about 2 for AUCunbound. The increased locomotor activity in offspring may indicate a potential risk for the foetus. In addition, because of the findings in pups, a risk for the nursing newborn/infant cannot be excluded.
Female fertility
Finerenone caused reduced female fertility (decreased number of corpora lutea and implantation sites) as well as signs of early embryonic toxicity (increased post-implantational loss and decreased number of viable foetuses) at about 21 times the human AUCunbound. In addition, reduced ovarian weights were found at about 17 times the human AUCunbound. No effects on female fertility and early embryonic development were found at 10 times the human AUCunbound. Therefore, the findings in female rats are of little clinical relevance (see section 4.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.