LEQEMBI Solution for injection Ref.[107292] Active ingredients: Lecanemab
Source: FDA, National Drug Code (US) Revision Year: 2023
12.1. Mechanism of Action
Lecanemab-irmb is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid beta. The accumulation of amyloid beta plaques in the brain is a defining pathophysiological feature of Alzheimer’s disease. LEQEMBI reduces amyloid beta plaques, as evaluated in Study 1 and Study 2 [see Clinical Studies (14)].
12.2. Pharmacodynamics
Effect of LEQEMBI on Amyloid Beta Pathology
The effect of LEQEMBI on amyloid beta plaque levels in the brain was evaluated using Positron Emission Tomography (PET) imaging. The PET signal was quantified using the both the Standard Uptake Value Ratio (SUVR) and Centiloid scale to estimate levels of amyloid beta plaque in composites of brain areas expected to be widely affected by Alzheimer’s disease pathology (frontal, parietal, lateral temporal, sensorimotor, and anterior and posterior cingulate cortices), compared to a brain region expected to be spared of such pathology (cerebellum).
LEQEMBI reduced amyloid beta plaque in a dose- and time-dependent manner in the dose-ranging study (Study 1) and in a time-dependent manner in single-dosing regimen study (Study 2) compared with placebo [see Clinical Studies (14)].
In Study 1, treatment with LEQEMBI 10 mg/kg every two weeks reduced amyloid beta plaque levels in the brain, producing reductions in PET SUVR compared to placebo at both Weeks 53 and 79 (p<0.0001). The magnitude of the reduction was time- and dose-dependent.
During an off-treatment period in Study 1 (range from 9 to 59 months; mean of 24 months), SUVR and Centiloid values began to increase with a mean rate of increase of 2.6 Centiloids/year, however, treatment difference relative to placebo at the end of the double-blind, placebo-controlled period in Study 1 was maintained.
In Study 2, treatment with LEQEMBI 10 mg/kg every two weeks reduced amyloid beta plaque levels in the brain, producing reductions compared to placebo starting at Week 13 and continuing through Week 79 (p<0.0001).
An increase in plasma Aβ42/40 ratio (Table 6) and CSF Aβ[1-42] was observed with LEQEMBI 10 mg/kg every two weeks dosing compared to placebo.
Effect of LEQEMBI on Tau Pathophysiology
A reduction in plasma p-tau181 (Table 6), CSF p-tau181, and CSF t-tau was observed with LEQEMBI 10 mg/kg every two weeks compared to placebo.
Table 6. Effect of LEQEMBI on Plasma Aβ42/40 and Plasma p-tau181 in Study 1 and Study 2:
| Biomarker Endpoints | Study 1 | Study 2 | ||
|---|---|---|---|---|
| LEQEMBI 10 mg/kg Every Two Weeks | Placebo | LEQEMBI 10 mg/kg Every Two Weeks | Placebo | |
| Plasma Aβ42/402 | N=43 | N=88 | N=797 | N=805 |
| Mean baseline | 0.0842 | 0.0855 | 0.088 | 0.088 |
| Adjusted mean change from baseline at Month 183 | 0.0075 | 0.0021 | 0.008 | 0.001 |
| Difference from placebo | 0.0054 (p=0.0036)1 | 0.007 (p<0.0001)1 | ||
| Plasma p-tau181 (pg/mL)2 | N=84 | N=179 | N=746 | N=752 |
| Mean baseline | 4.6474 | 4.435 | 3.696 | 3.740 |
| Adjusted mean change from baseline at Month 183 | -1.1127 | 0.0832 | -0.575 | 0.201 |
| Difference from placebo | -1.1960 (p<0.0001)1 | -0.776 (p<0.0001)1 | ||
N is the number of patients with baseline value.
1 P-values were not statistically controlled for multiple comparisons.
2 Results should be interpreted with caution due to uncertainties in bioanalysis.
3 Month 18 represents Week 79 in Study 1 and Week 77 in Study 2
A substudy was conducted in Study 2 to evaluate the effect of LEQEMBI on neurofibrillary tangles composed of tau protein using PET imaging ( 18F-MK6240 tracer). The PET signal was quantified using the SUVR method to estimate brain levels of tau in brain regions expected to be affected by Alzheimer’s disease pathology (whole cortical gray matter, meta-temporal, frontal, cingulate, parietal, occipital, medial temporal, and temporal) in the study population compared to a brain region expected to be spared of such pathology (cerebellum). The adjusted mean change from baseline in tau PET SUVR, relative to placebo, was in favor of LEQEMBI in the medial temporal (p<0.01), meta temporal (p<0.05), and temporal (p<0.05) regions. No statistically significant differences were observed for the whole cortical gray matter, frontal, cingulate, parietal, or occipital regions.
Exposure-Response Relationships
Model based exposure-response analyses demonstrated that higher exposures to lecanemab-irmb were associated with greater reduction in clinical decline on Clinical Dementia Rating scale Sum of Boxes (CDR-SB) and Alzheimer Disease Assessment Scale – Cognitive Subscale 14 (ADAS-Cog14). In addition, higher exposures to lecanemab-irmb were associated with greater reduction in amyloid beta plaque. An association between reduction in amyloid beta plaque and clinical decline on CDR-SB and ADAS-Cog14 was also observed.
Higher exposures to lecanemab-irmb were also associated with greater increase in plasma Aβ42/40 ratio and greater reduction in plasma p-tau181.
12.3. Pharmacokinetics
Steady-state concentrations of lecanemab-irmb were reached after 6 weeks of 10 mg/kg administered every 2 weeks and systemic accumulation was 1.4-fold. The peak concentration (Cmax) and area under the plasma concentration versus time curve (AUC) of lecanemab-irmb increased dose proportionally in the dose range of 0.3 to 15 mg/kg following single dose.
Distribution
The mean value (95% CI) for central volume of distribution at steady-state is 3.24 (3.18-3.30) L.
Elimination
Lecanemab-irmb is degraded by proteolytic enzymes in the same manner as endogenous IgGs. The clearance of lecanemab-irmb (95% CI) is 0.370 (0.353-0.384) L/day. The terminal half-life is 5 to 7 days.
Specific Populations
Sex, body weight, and albumin were found to impact exposure to lecanemab-irmb. However, none of these covariates were found to be clinically significant.
Patients with Renal or Hepatic Impairment
No clinical studies were conducted to evaluate the pharmacokinetics of lecanemab-irmb in patients with renal or hepatic impairment. Lecanemab-irmb is degraded by proteolytic enzymes and is not expected to undergo renal elimination or metabolism by hepatic enzymes.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted.
Mutagenesis
Genotoxicity studies have not been conducted.
Impairment of Fertility
No studies in animals have been conducted to assess the effects of lecanemab-irmb on male or female fertility. No adverse effects on male or female reproductive organs were observed in a 39-week intravenous toxicity study in monkeys administered lecanemab-irmb weekly at doses up to 100 mg/kg. The highest dose tested was associated with plasma exposures (Cave) approximately 27 times that in humans at the recommended human dose (10 mg/kg every two weeks).
14. Clinical Studies
The efficacy of LEQEMBI was evaluated in two double-blind, placebo-controlled, parallel-group, randomized studies (Study 1, NCT01767311; Study 2 NCT03887455) in patients with Alzheimer’s disease (patients with confirmed presence of amyloid pathology and mild cognitive impairment [64% of patients in Study 1; 62% of patients in Study 2] or mild dementia stage of disease [36% of patients in Study 1; 38% of patients in Study 2], consistent with Stage 3 and Stage 4 Alzheimer’s disease). In both studies, patients were enrolled with a Clinical Dementia Rating (CDR) global score of 0.5 or 1.0 and a Memory Box score of 0.5 or greater. All patients had a Mini-Mental State Examination (MMSE) score of ≥22 and ≤30, and had objective impairment in episodic memory as indicated by at least 1 standard deviation below age-adjusted mean in the Wechsler-Memory Scale-IV Logical Memory II (subscale) (WMS-IV LMII). Patients were enrolled with or without concomitant approved therapies (cholinesterase inhibitors and the N-methyl-D-aspartate antagonist memantine) for Alzheimer’s disease. Patients in each study could enroll in an optional, long-term extension.
Study 1
In Study 1, 856 patients were randomized to receive one of 5 doses (161 of which were randomized to the recommended dosing regimen of 10 mg/kg every two weeks) of LEQEMBI or placebo (n=247). Of the total number of patients randomized, 71.4% were ApoE ε4 carriers and 28.6% were ApoE ε4 non-carriers. During the study the protocol was amended to no longer randomize ApoE ε4 carriers to the 10 mg/kg every two weeks dose arm. ApoE ε4 carriers who had been receiving LEQEMBI 10 mg/kg every two weeks for 6 months or less were discontinued from study drug. As a result, in the LEQEMBI 10 mg/kg every two weeks arm, 30.3% of patients were ApoE ε4 carriers and 69.7% were ApoE ε4 non-carriers. At baseline, the mean age of randomized patients was 71 years, with a range of 50 to 90 years. Fifty percent of patients were male and 90% were White.
In Study 1, a subgroup of 315 patients were enrolled in the amyloid PET substudy; of these, 277 were evaluated at Week 79. Results from the amyloid beta PET substudy are described in Figure 1 and Table 7. Plasma biomarkers are described in Table 5.
Figure 1. Reduction in Brain Amyloid Beta Plaque (Adjusted Mean Change from Baseline in Amyloid Beta PET Composite, SUVR and Centiloids) in Study 1:
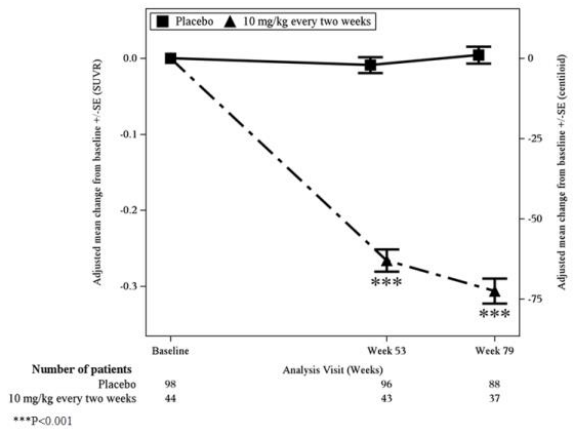
Table 7. Results for Amyloid Beta PET in Study 1:
| Biomarker Endpoints | LEQEMBI 10 mg/kg Every Two Weeks | Placebo |
|---|---|---|
| Amyloid Beta PET Composite SUVR | N=44 | N=98 |
| Mean baseline | 1.373 | 1.402 |
| Adjusted mean change from baseline at Week 79 | -0.306 | 0.004 |
| Difference from placebo | -0.310 (p<0.001) 1 | |
| Amyloid Beta PET Centiloid | N=44 | N=98 |
| Mean baseline | 78.0 | 84.8 |
| Adjusted mean change from baseline at Week 79 | -72.5 | 1.0 |
| Difference from placebo | -73.5 (p<0.001) 1 |
N is the number of patients with baseline value.
1 P-values were not statistically controlled for multiple comparisons.
The primary endpoint was change from baseline on a weighted composite score consisting of selected items from the Clinical Dementia Rating scale Sum of Boxes (CDR-SB), MMSE, and Alzheimer Disease Assessment Scale – Cognitive Subscale 14 (ADAS-Cog 14) at Week 53. LEQEMBI had a 64% likelihood of 25% or greater slowing of progression on the primary endpoint relative to placebo at Week 53, which did not meet the prespecified success criterion of 80%.
Key secondary efficacy endpoints included the change from baseline in amyloid PET SUVR composite at Week 79 and change from baseline in the CDR-SB and ADAS-Cog14 at Week 79. Results for clinical assessments showed less change from baseline in CDR-SB and ADAS-Cog 14 scores at Week 79 in the LEQEMBI group than in patients on placebo (CDR-SB: -0.40 [26%], 90% CI [-0.82, 0.03]; ADAS-Cog 14: -2.31 [47%], 90% CI [-3.91, -0.72].
After the 79-week double-blind, placebo-controlled period of Study 1, patients could enroll in an open-label extension period for up to 260 weeks, which was initiated after a gap period (range 9 to 59 months; mean 24 months) off treatment.
Study 2
In Study 2, 1795 patients were enrolled and randomized 1:1 to receive LEQEMBI 10 mg/kg or placebo once every 2 weeks. Of the total number of patients randomized, 69% were ApoE ε4 carriers and 31% were ApoE ε4 non-carriers. Overall median age of patients was 72 years, with a range of 50 to 90 years. Fifty-two percent were women, and 1381 (77%) were White, 303 (17%) were Asian, and 47 (3%) were Black.
The randomization was stratified according to clinical subgroup (mild cognitive impairment or mild dementia stage of the disease); the presence or absence of concomitant approved therapies for Alzheimer’s disease at baseline (cholinesterase inhibitors and the N-methyl-D-aspartate antagonist memantine); ApoE ε4 carrier status; and geographical region.
The primary efficacy outcome was change from baseline at 18 months in the CDR-SB. Key secondary endpoints included change from baseline at 18 months for the following measures: amyloid Positron Emission Tomography (PET) using Centiloids, ADAS-Cog14, and Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL).
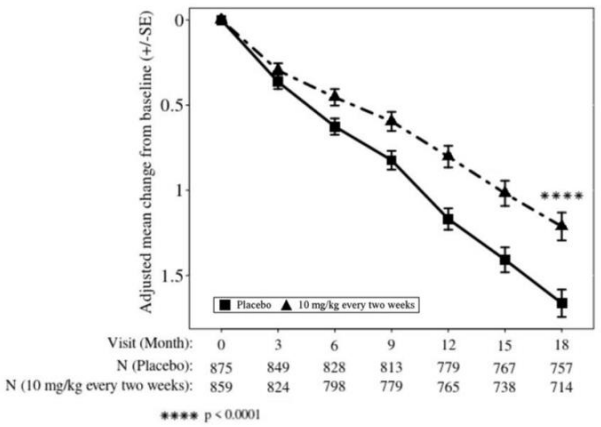
LEQEMBI treatment met the primary endpoint and reduced clinical decline on the global cognitive and functional scale, CDR-SB, compared with placebo at 18 months (-0.45 [-27%], p<0.0001).
Statistically significant differences (p<0.01) between treatment groups were also seen in the results for ADAS-Cog14 and ADCS MCI-ADL at 18 months as presented in Table 8.
Both ApoE ε4 carriers and ApoE ε4 noncarriers showed statistically significant treatment differences for the primary endpoint and all secondary endpoints. In an exploratory subgroup analysis of ApoE ε4 homozygotes, which represented 15% of the trial population, a treatment effect was not observed with LEQEMBI treatment on the primary endpoint, CDR-SB, compared to placebo, although treatment effects that favored LEQEMBI were observed for the secondary clinical endpoints, ADAS-Cog 14 and ADCS MCI-ADL. Treatment effects on disease-relevant biomarkers (amyloid beta PET, plasma Aβ42/40 ratio, plasma p-tau 181) also favored LEQEMBI in the ApoE ε4 homozygous subgroup.
Starting at six months, across all time points, LEQEMBI treatment showed statistically significant changes in the primary and all key secondary endpoints from baseline compared to placebo; see Figure 2.
Table 8. Results for CDR-SB, ADAS-Cog14, and ADCS MCI-ADL in Study 2:
| Clinical Endpoints | LEQEMBI 10 mg/kg Every Two Weeks | Placebo |
|---|---|---|
| CDR-SB | N=859 | N=875 |
| Mean baseline | 3.17 | 3.22 |
| Adjusted mean change from baseline at 18 months (%) Difference from placebo | 1.21 -0.45 (-27%) (p<0.0001) | 1.66 |
| ADAS-Cog14 | N=854 | N=872 |
| Mean baseline | 24.45 | 24.37 |
| Adjusted mean change from baseline at 18 months (%) Difference from placebo | 4.140 -1.442 (-26%) (p=0.00065) | 5.581 |
| ADCS MCI-ADL | N=783 | N=796 |
| Mean baseline | 41.2 | 40.9 |
| Adjusted mean change from baseline at 18 months Difference from placebo | -3.5 (-37%) 2.0 (p<0.0001) | -5.5 |
Figure 2: Adjusted Mean Change from Baseline in CDR-SB in Study 2:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.