LIBTAYO Concentrate for solution for infusion Ref.[8754] Active ingredients: Cemiplimab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Regeneron Ireland Designated Activity Company (DAC), One Warrington Place, Dublin 2, D02 HH27, Ireland
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, PD-1/PD-L1 (Programmed cell death protein 1/death ligand 1) inhibitors
ATC code: L01FF06
Mechanism of action
Cemiplimab is a fully human immunoglobulin G4 (IgG4) monoclonal antibody that binds to the programmed cell death-1 (PD-1) receptor and blocks its interaction with its ligands PD-L1 and PD-L2. Engagement of PD-1 with its ligands PD-L1 and PD-L2, which are expressed by antigen presenting cells and may be expressed by tumour cells and/or other cells in the tumour microenvironment, results in inhibition of T cell function such as proliferation, cytokine secretion, and cytotoxic activity. Cemiplimab potentiates T cell responses, including anti-tumour responses, through blockade of PD-1 binding to PD-L1 and PD-L2 ligands.
Clinical efficacy and safety
CSCC
The efficacy and safety of cemiplimab in patients with mCSCC (nodal or distant) or laCSCC who were not candidates for curative surgery or curative radiation were studied in clinical trial R2810-ONC-1540 (Study 1540). Study 1540 was a phase 2, open-label, multi-centre study that enrolled 193 patients with mCSCC or laCSCC in Groups 1 to 3 with a combined median follow-up time of 15.7 months total. Median duration of follow-up was 18.5 months for the mCSCC 3 mg/kg every 2 weeks (Q2W) group (Group 1), 15.5 months for the laCSCC 3 mg/kg Q2W group (Group 2), 17.3 months for the mCSCC 350 mg Q3W group (Group 3). In an additional cohort of 165 advanced CSCC patients (mCSCC and laCSCC) dosed at 350 mg Q3W, the median duration of follow-up was 8.7 months (Group 6).
Patients with any of the following were excluded: autoimmune disease that required systemic therapy with immunosuppressant agents within 5 years; history of solid organ transplant; history of pneumonitis within the last 5 years; prior treatment with anti-PD-1/PD-L1 or other immune checkpoint inhibitor therapy; active infection requiring therapy, including known infection with human immunodeficiency virus, or active infection with hepatitis B or hepatitis C virus; chronic lymphocytic leukaemia (CLL); brain metastases or Eastern Cooperative Oncology Group (ECOG) performance score (PS) ≥2.
In Study 1540, patients received cemiplimab intravenously (IV) until progression of disease, unacceptable toxicity or completion of planned treatment [3 mg/kg Q2W for 96 weeks (Groups 1 and 2) or 350 mg Q3W for 54 weeks (Group 3)]. If patients with locally advanced disease showed sufficient response to treatment, surgery with curative intent was permitted. Tumour response assessments were performed every 8 or 9 weeks (for patients receiving 3 mg/kg Q2W or 350 mg Q3W, respectively). The primary efficacy endpoint of Study 1540 was confirmed objective response rate (ORR), as assessed by independent central review (ICR). For patients with mCSCC without externally visible target lesions, ORR was determined by Response Evaluation Criteria in Solid Tumours (RECIST 1.1). For patients with externally visible target lesions (laCSCC and mCSCC), ORR was determined by a composite endpoint that integrated ICR assessments of radiologic data (RECIST 1.1) and digital medical photography (WHO criteria). The key secondary endpoint was duration of response (DOR) by ICR. Other secondary endpoints included ORR and DOR by investigator assessment (IA), progression-free survival (PFS) by ICR and IA, overall survival (OS), complete response rate (CR) by ICR, and change in scores in patient reported outcomes on the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire (EORTC QLQ-C30).
In the efficacy analysis of 193 patients with advanced CSCC from Study 1540 Groups 1 to 3, 115 had mCSCC and 78 had laCSCC. The median age was 72 years (range: 38 to 96): Seventy-eight (40.4%) patients were 75 years or older, 66 patients (34.2%) were 65 to less than 75 years, and 49 patients (25.4%) were less than 65 years. A total of 161 (83.4 ) patients were male, and 187 (96.9) patients were White; the ECOG PS was 0 (44.6%) and 1 (55.4%). Thirty-three and 7/10 percent (33.7%) of patients had received at least 1 prior anti-cancer systemic therapy, 81.3% of patients had received prior cancer related surgery, and 67.9% of patients had received prior radiotherapy. Among patients with mCSCC, 76.5% had distant metastases, and 22.6% had only nodal metastases.
Efficacy results based on the final analysis of Study 1540 Groups 1 to 3 are presented in Table 3.
Table 3. Efficacy results – Study 1540 – metastatic CSCC by dosing group, locally advanced CSCC:
| Efficacy endpoints | mCSCC cemiplimab: 3 mg/kg every 2 weeks (Group 1) (N=59) | laCSCC cemiplimab: 3 mg/kg every 2 weeks (Group 2) (N=78) | mCSCC cemiplimab: 350 mg every 3 weeks (Group 3) (N=56) |
|---|---|---|---|
| ICR | ICR | ICR | |
| Confirmed objective response rate (ORR)a | |||
| ORR | 50.8% | 44.9% | 46.4% |
| 95% CI for ORR | (37.5, 64.1) | (33.6, 56.6) | (33.0, 60.3) |
| Complete response (CR)b | 20.3% | 12.8% | 19.6% |
| Partial response (PR) | 30.5% | 32.1% | 26.8% |
| Stable disease (SD) | 15.3% | 34.6% | 14.3% |
| Progressive disease (PD) | 16.9% | 12.8% | 25.0% |
| Duration of response (DOR) | |||
| Medianc (months) (95% CI) | NR (20.7, NE) | 41.9 (20.5, 54.6) | 41.3 (40.8, 46.3) |
| Range (months) | 2.8-38.9 | 1.9-54.6 | 4.2-46.3 |
| Patients with DOR ≥6 months, % | 93.3% | 88.6% | 96.2% |
| Time to response (TTR) | |||
| Median (months) range (min:max) | 1.9 (1.7: 21.8) | 2.1 (1.8: 8.8) | 2.1 (2.0: 22.8) |
| Progression-free survival (PFS)a,c | |||
| 6 months (95% CI) | 66.4% (52.5, 77.1) | 72.4% (60.1, 81.5) | 60.7% (46.7, 72.1) |
| 12 months (95% CI) | 53.8% (40.0, 65.8) | 60.8% (47.8, 71.5) | 53.4% (39.5, 65.4) |
| Overall survival (OS)a,c | |||
| 12 months (95% CI) | 81.3% (68.7, 89.2) | 91.8% (82.6, 96.2) | 72.5% (58.6, 82.5) |
CI: Confidence interval; ICR: Independent Central Review; NR: Not reached; NE: Not evaluable.
a In Groups 1, 2, and 3, median durations of follow-up were 18.5, 15.5, and 17.3 months, respectively.
b Only includes patients with complete healing of prior cutaneous involvement; laCSCC patients in Study 1540 required biopsy to confirm CR.
c Based on Kaplan Meier estimates.
Efficacy and PD-L1 status
Clinical activity was observed regardless of tumour PD-L1 expression status.
BCC
The efficacy and safety of cemiplimab in patients with laBCC or mBCC who had progressed on HHI therapy, were intolerant of prior HHI therapy, or had no better than SD after 9 months on HHI therapy (exclusive of treatment breaks), were evaluated in Study 1620, an open-label, multi-centre, non-randomised study. The study excluded patients with autoimmune disease that required systemic therapy with immunosuppressant agents within 5 years; history of solid organ transplant; prior treatment with anti-PD-1/PD-L1 therapy or other immune checkpoint inhibitor therapy; infection with HIV, hepatitis B or hepatitis C; or ECOG performance score (PS) ≥2.
Patients received cemiplimab 350 mg intravenously (IV) every 3 weeks for 5 cycles of 9 weeks followed by 4 cycles of 12 weeks up to 93 weeks of treatment. Treatment continued until disease progression, unacceptable toxicity or completion of planned treatment. Tumour assessments were performed every 9 weeks during cycles 1 to 5 and every 12 weeks during cycles 6 to 9. The major efficacy endpoints were confirmed ORR and DOR as assessed by ICR. Secondary efficacy outcomes included ORR and DOR by IA, PFS, OS, CR by ICR, and time to response. For patients with mBCC without externally visible target lesions, ORR was determined by RECIST 1.1. For patients with externally visible target lesions (laBCC and mBCC), ORR was determined by a composite endpoint that integrated ICR assessments of radiologic data (RECIST 1.1) and digital medical photography (WHO criteria).
A total of 138 patients with advanced BCC were included in the efficacy analysis of Study 1620, 84 patients with laBCC and 54 patients with mBCC.
In the laBCC group, the median age was 70.0 years (range: 42 to 89): 31 (37%) patients were <65 years old and 53 (63%) were 65 years or older. A total of 56 (67%) were male and 57 (68%) were White; the ECOG PS was 0 (61%) and 1 (39%); Eighty-three per cent (83%) of patients had received at least 1 prior cancer-related surgery and 35% of patients had >3 prior cancer-related surgeries (median: 3.0 surgeries, range: 1 to 43); 50% of patients had received at least 1 prior anti-cancer radiotherapy (RT) (median: 1.0 RT, range: 1 to 6).
In the mBCC group, the median age was 63.5 years (range: 38 to 90): 27 (50%) patients were <65 years old and 27 (50%) were 65 years or older. A total of 38 (70%) were male and 47 (87%) were White; the ECOG PS was 0 (67%) and 1 (33%); Eighty-five per cent (85%) of patients had received at least 1 prior cancer-related surgery and 28% of patients had >3 prior cancer-related surgeries (median: 2.0 surgeries, range: 1 to 8); 59% of patients had received at least 1 prior anti-cancer radiotherapy (RT) (median: 1.0 RT, range: 1 to 4).
All 138 patients were previously treated with a HHI, and 12% (16/138) of patients were previously treated with both vismodegib and sonidegib (as separate lines of therapy). Of the 84 laBCC patients, 71% (60/84) of patients discontinued HHI therapy due to disease progression, 38% (32/84) of patients discontinued HHI therapy due to intolerance and 2% (2/84) discontinued solely due to lack of response. Of the 54 mBCC patients, 76% (41/54) of patients discontinued HHI therapy due to disease progression, 33% (18/54) of patients discontinued HHI therapy due to intolerance, and 6% (3/54) discontinued solely due to lack of response. Investigators could select more than one reason for discontinuation of prior HHI therapy for an individual patient.
Efficacy results are presented in Table 4.
Table 4. Efficacy results for Study 1620 in locally advanced and metastatic basal cell carcinoma:
| Efficacy endpoints | laBCC cemiplimab 350 mg every 3 weeks | mBCC cemiplimab 350 mg every 3 weeks |
|---|---|---|
| N=84 | N=54 | |
| ICR | ICR | |
| Best overall response (BOR)a,b,c | ||
| Objective response rate (ORR: CR+ PR) (95% CI) | 27 (32.1%) (22.4, 43.2) | 12 (22.2%) (12.0, 35.6) |
| Complete response (CR) rated (95% CI) | 6 (7.1%) (2.7, 14.9) | 1 (1.9%) (0.0, 9.9) |
| Partial response (PR) rate | 21 (25.0%) | 11 (20.4%) |
| Progressive disease (PD) rate | 9 (10.7%) | 16 (29.6%) |
| Duration of response (DOR) | N=27 responders | N=12 responders |
| Mediane (months) (95% CI) | NR (15.5, NE) | 16.7 (9.8, NE) |
| Range (observed) (months) | 2.1 – 36.8+ | 9.0 – 25.8+ |
| Patients with DOR ≥6 months, e (95 CI) | 88.5% (68.4, 96.1) | 100.0% (100, 100) |
| Time to response (TTR) | N=27 responders | N=12 responders |
| Median (months) (Range) | 4.3 (2.1 – 21.4) | 3.1 (2.0 – 10.5) |
CI: Confidence interval; +: Denotes ongoing at last assessment; ICR: Independent Central Review; NR: Not reached; NE: Not evaluable
a Median duration of follow-up: laBCC: 15.9 months, mBCC: 8.4 months.
b Includes 2 laBCC patients who met the inclusion criteria solely on the basis of “No better than stable disease (SD) after 9 months on HHI therapy”. BOR results by ICR were SD for 1 patient and NE for 1 patient.
c Includes 3 mBCC patients who met the inclusion criteria solely on the basis of “No better than SD after 9 months on HHI therapy”. BOR results by ICR were PR for 1 patient and SD for 2 patients.
d Locally advanced BCC patients in Study 1620 required biopsy to confirm complete response.
e Based on Kaplan Meier estimates.
Efficacy and PD-L1 status
Clinical activity was observed regardless of tumour PD-L1 expression status.
NSCLC
First-line treatment of NSCLC with cemiplimab as monotherapy
The efficacy and safety of cemiplimab compared with platinum-doublet chemotherapy in patients with locally advanced NSCLC who were not candidates for definitive chemoradiation, or with metastatic NSCLC who had tumour PD-L1 expression ≥50% using the PD-L1 IHC 22C3 pharmDx assay were evaluated in Study 1624, a randomised, open-label, multi-centre study.
A total of 710 patients were enrolled.
The study excluded patients with EGFR, ALK or ROS1 genomic tumour aberrations, ECOG performance score (PS) ≥2, medical conditions that required systemic immunosuppression, uncontrolled infection with hepatitis B (HBV) or hepatitis C (HCV) or human immunodeficiency virus (HIV), history of interstitial lung disease, who were never smokers or who had an autoimmune disease that required systemic therapy within 2 years of treatment. Treatment of brain metastases was permitted, and patients could be enrolled if they had been adequately treated and had neurologically returned to baseline for at least 2 weeks prior to randomisation. Radiological confirmation of stability or response was not required.
Randomisation was stratified by histology (non-squamous vs squamous) and geographic region (Europe, Asia, or Rest of World). Patients were randomised (1:1) to receive cemiplimab 350 mg intravenously (IV) every 3 weeks for up to 108 weeks or investigator’s choice of the following platinum-doublet chemotherapy regimens for 4 to 6 cycles: paclitaxel + cisplatin or carboplatin; gemcitabine + cisplatin or carboplatin; or pemetrexed + cisplatin or carboplatin followed by optional pemetrexed maintenance (This regimen was not recommended for patients with squamous NSCLC).
Treatment with cemiplimab continued until RECIST 1.1-defined progressive disease, unacceptable toxicity, or up to 108 weeks. Patients who experienced independent review committee (IRC)-assessed RECIST 1.1-defined progressive disease on cemiplimab therapy were permitted to continue treatment with cemiplimab with an addition of 4 cycles of histology-specific chemotherapy until further progression was observed. Patients who experienced IRC-assessed RECIST 1.1-defined progressive disease on chemotherapy treatment were permitted to receive cemiplimab treatment until further progression, unacceptable toxicity or up to 108 weeks. Of the 203 patients randomised to receive chemotherapy who had IRC-assessed RECIST 1.1-defined disease progression, 150 (73.9%) patients crossed over to treatment with cemiplimab. Assessment of tumour status was performed every 9 weeks. The primary efficacy endpoints were overall survival (OS) and progression-free survival (PFS) as assessed by blinded IRC using RECIST 1.1. A key secondary endpoint was objective response rate (ORR).
Among the 710 patients, baseline characteristics were: median age 63 years (45% were 65 or older), 85% male, 86% White, an ECOG performance score 0 and 1 in 27% and 73% respectively, and 12% with history of brain metastasis. Disease characteristics were locally advanced (16%), metastatic (84%), squamous (44%) and non-squamous (56%).
The study showed statistically significant improvement in OS for patients randomised to cemiplimab as compared with chemotherapy.
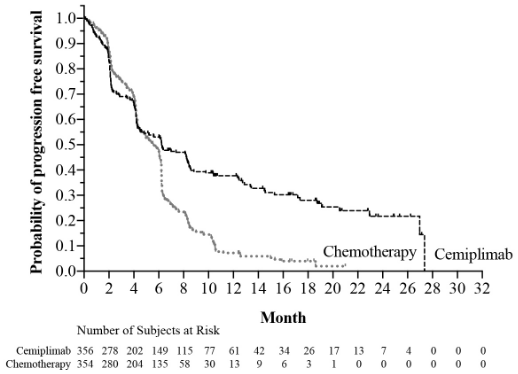
Efficacy results are presented in Table 5, Figure 1 and Figure 2.
Table 5. Efficacy results for Study 1624 in non-small cell lung cancer:
| Efficacy endpointsa | Cemiplimab 350 mg every 3 weeks N=356 | Chemotherapy N=354 |
|---|---|---|
| Overall survival (OS) | ||
| Deaths n (%) | 108 (30.3) | 141 (39.8) |
| Median in months (95% CI)b | 22.1 (17.7, NE) | 14.3 (11.7, 19.2) |
| Hazard ratio (95% CI)c | 0.68 (0.53, 0.87) | |
| p-Valued | 0.0022 | |
| OS rate at 12 months (95% CI)b | 70% (64, 75) | 56% (49, 62) |
| Progression-free survival (PFS) | ||
| Events n (%) | 201 (56.5) | 262 (74.0) |
| Median in months (95% CI)b | 6.2 (4.5, 8.3) | 5.6 (4.5, 6.1) |
| Hazard ratio (95% CI)c | 0.59 (0.49, 0.72) | |
| PFS rate at 12 months (95% CI)b | 38% (32, 44) | 7% (4, 11) |
| Objective response rate (%)e | ||
| ORR (95% CI) | 36.5 (31.5, 41.8) | 20.6 (16.5, 25.2) |
| Complete response (CR) rate | 3.1 | 0.8 |
| Partial response (PR) rate | 33.4 | 19.8 |
| Duration of response | N=130 responders | N=73 responders |
| Median (months)b | 21.0 | 6.0 |
| Range (months) | (1.9 , 23.3) | (1.3+, 16.5+) |
| Patients with observed DOR ≥6 months, % | 69% | 41% |
CI: Confidence interval; NE: Not evaluable; +: Ongoing response
a Median duration of follow-up: cemiplimab: 13.1 months; chemotherapy: 13.1 months
b Based on Kaplan-Meier estimates
c Based on stratified proportional hazards model
d Based on a two-sided p-value
e Based on Clopper-Pearson exact confidence interval
Figure 1. OS in Study 1624 in NSCLC:
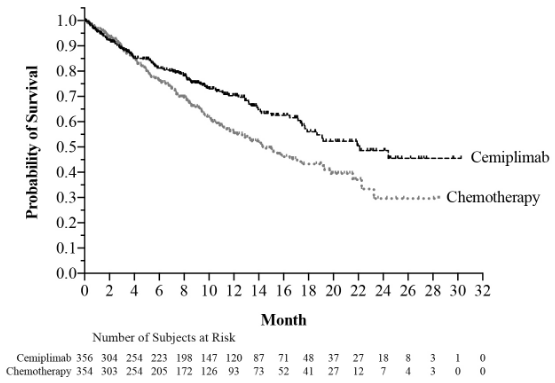
Figure 2. PFS in Study 1624 in NSCLC:
First-line treatment of NSCLC with cemiplimab in combination with platinum-based chemotherapy
The efficacy and safety of cemiplimab in combination with platinum-based chemotherapy were evaluated in Study 16113, a randomised, multi-centre, double-blind, active-controlled trial in 466 patients with locally advanced NSCLC who were not candidates for definitive chemoradiation, or with metastatic NSCLC, regardless of tumour PD-L1 expression status and who had not previously received systemic treatment for metastatic NSCLC. Testing for genomic tumour aberrations other than EGFR, ALK or ROS1 was not mandatory for enrolment in Study 16113.
Patients with EGFR, ALK or ROS1 genomic tumour aberrations; a medical condition that required systemic immunosuppression; active infection with hepatitis B (HBV) or hepatitis C (HCV), uncontrolled human immunodeficiency disease (HIV), or ongoing or recent autoimmune disease that required systemic therapy were ineligible. Patients with a history of brain metastases were eligible if they had been adequately treated and had neurologically returned to baseline for at least 2 weeks prior to randomisation. Radiological confirmation of stability or response was not required.
Randomisation was stratified by histology (non-squamous vs squamous) and PD-L1 expression (<1% versus 1% to 49% versus ≥50%) according to the VENTANA PD-L1 (SP263) assay. Patients were randomised (2:1) to receive either cemiplimab 350 mg intravenously (IV) every 3 weeks for 108 weeks plus platinum-based chemotherapy every 3 weeks for 4 cycles or placebo intravenously (IV) every 3 weeks for 108 weeks plus platinum-based chemotherapy every 3 weeks for 4 cycles.
Treatment with cemiplimab or placebo was continued until RECIST 1.1-defined progressive disease, unacceptable toxicity, or up to 108 weeks. Treatment with chemotherapy was given for 4 cycles followed by maintenance of pemetrexed as clinically indicated or until RECIST 1.1-defined progressive disease or unacceptable toxicity. Chemotherapy in Study 16113 consisted of carboplatin or cisplatin combined with paclitaxel or pemetrexed with mandatory maintenance for pemetrexed regimens. Assessment of tumour status was performed every 9 weeks beginning at week 9 during year 1 and every 12 weeks beginning at week 55 during year 2. The primary efficacy endpoint was overall survival (OS). Key secondary endpoints as assessed by blinded IRC using RECIST 1.1, were progression-free survival (PFS), and objective response rate (ORR).
Among the 466 patients, 327 (70%) had tumours expressing PD-L1 (in ≥1% of tumour cells). Of these, 217 patients were in the cemiplimab and chemotherapy group and 110 patients were in the placebo and chemotherapy group. The baseline characteristics of the 327 patients with tumours expressing PD-L1 in ≥1% of tumour cells were: median age 62 years (38% were 65 or older), 83% male, 87% White, an ECOG performance score 0 and 1 in 16% and 83% respectively, and 6% with history of brain metastasis; 51% of patients were current smokers, 34% were past smokers and 15% had never smoked (less than 100 cigarettes a lifetime). Disease characteristics were locally advanced (14%), metastatic (86%), squamous histology (45%), and non-squamous histology (55%).
At the primary analysis in the overall population with a median follow-up time of 16.4 months, the study showed a statistically significant improvement in OS for patients randomised to cemiplimab in combination with chemotherapy compared with placebo in combination with chemotherapy.
Efficacy results in patients whose tumours expressed PD-L1 ≥1% are presented in Table 6, Figure 3, and Figure 4.
Table 6. Efficacy results for Study 16113 in non-small cell lung cancer (patients with PD-L1 expression ≥1%)a:
| Endpointsa | cemiplimab and chemotherapy N=217 | placebo and chemotherapy N=110 |
|---|---|---|
| Overall Survival (OS) | ||
| Deaths, n (%) | 78 (35.9) | 55 (50.0) |
| Median in months (95% CI)b | 21.9 (17.3, NE) | 12.6 (10.3, 16.4) |
| Hazard ratio (95% CI)c | 0.55 (0.39, 0.78) | |
| Progression-free Survival (PFS) | ||
| Events, n (%) | 134 (61.8) | 86 (78.2) |
| Median in months (95% CI)b | 8.5 (6.7, 10.7) | 5.5 (4.3, 6.2) |
| Hazard ratio (95% CI)c | 0.48 (0.36, 0.63) | |
| Objective Response Rate (ORR) (%) | ||
| ORR (95% CI)d | 47.9 (41.1, 54.8) | 22.7 (15.3, 31.7) |
| Complete response (CR) rate | 2.8 | 0 |
| Partial response (PR) rate | 45.2 | 22.7 |
| Duration of Response (DOR) | ||
| Median in monthsb (range) | 15.6 (1.7, 18.7+) | 4.9 (1.9, 18.8+) |
CI: confidence interval; NE: Not evaluable; +: Ongoing response (Data cutoff – Jun 14, 2021)
a Median duration of follow up: cemiplimab and chemotherapy: 15.9 months, placebo and chemotherapy: 16.1 months
b Based on Kaplan-Meier method
c Based on stratified proportional hazards model
d Clopper-Pearson exact confidence interval
At the time of the pre-specified final analysis, patients whose tumours expressed PD-L1 ≥1% randomised to cemiplimab in combination with chemotherapy, at a median duration of follow-up of 27.9 months, continued to show a clinically meaningful survival and progression free survival benefit compared to chemotherapy alone.
Figure 3. OS in Study 16113 in NSCLC (patients with PD-L1 expression ≥1%) – (Final analysis):
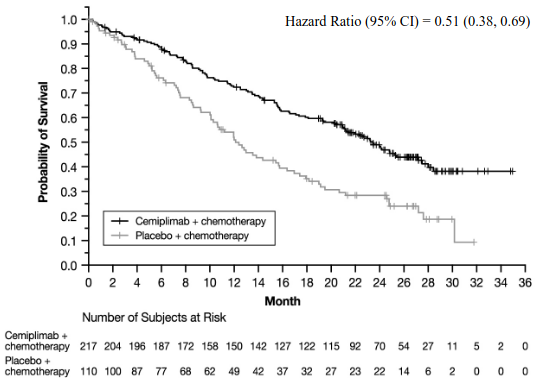
a Based on final OS analysis (Data cutoff Jun 14, 2022)
Figure 4. PFS in Study 16113 in NSCLC (patients with PD-L1 expression ≥ 1%) – (Final analysis)a:
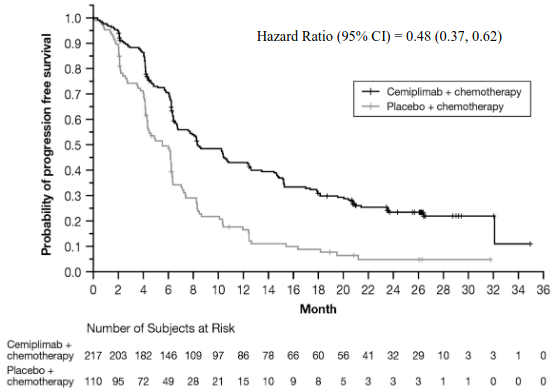
a Based on final PFS analysis (Data cutoff Jun 14, 2022)
Cervical Cancer
The efficacy and safety of cemiplimab were evaluated in patients with recurrent or metastatic cervical cancer whose tumours progressed on or after platinum-based chemotherapy, with or without bevacizumab in Study 1676, a randomised, open-label, multi-centre study. Patients were enrolled regardless of PD-L1 tumour expression status. The study excluded patients with autoimmune disease that required systemic therapy with immunosuppressant agents within 5 years and prior treatment with anti-PD-1/PD-L1 therapy.
The stratification factors for the efficacy analysis were geographic region (North America, Asia, Rest of World) and histology [squamous histology (SCC), adenocarcinoma/adenosquamous histologies (AC)]. Randomisation was also stratified by whether or not patients had received prior bevacizumab treatment and their ECOG performance status. Patients were randomised (1:1) to receive cemiplimab 350 mg intravenously every 3 weeks or investigator’s choice of intravenous chemotherapy among pemetrexed, topotecan, irinotecan, gemcitabine, or vinorelbine, for up to 96 weeks.
Treatment continued until disease progression, unacceptable toxicity, or completion of planned treatment. Tumour assessments were performed every 6 weeks for the first 24 weeks and every 12 weeks thereafter. The primary efficacy endpoint was OS in SCC followed by the total population. Secondary endpoints included PFS, ORR according to RECIST 1.1, and DOR by investigator assessment.
The median age was 51 years (22 to 87 years); 63% were White, 29% Asian, 3.5% Black; 49% received prior bevacizumab treatment, 47% had ECOG PS 0 and 53% had ECOG PS 1; 78% had SCC and 22% had AC, 94% had metastatic disease; 57% had 1 prior line of treatment in the recurrent or metastatic setting and 43% had > 1 prior line of treatment in the recurrent or metastatic setting. The median duration of follow-up for the primary analysis in the total population was 18.2 months.
Cemiplimab showed a statistically significant improvement in OS in both SCC and total population compared to chemotherapy.
Efficacy results are presented in Table 7, Figure 5, and Figure 6.
Table 7. Efficacy results for Study 1676 in cervical cancer:
| Squamous histology (SCC) (N=477) | Total population (N=608) | |||
|---|---|---|---|---|
| Efficacy endpoints | cemiplimab 350 mg every 3 weeks (n=239) | chemotherapy (n=238) | cemiplimab 350 mg every 3 weeks (n=304) | chemotherapy (n=304) |
| Overall survival (OS)a | ||||
| Deaths, n (%) | 143 (59.8%) | 161 (67.6%) | 184 (60.5%) | 211 (69.4%) |
| Median in months (95% CI)b | 11.1 (9.2, 13.4) | 8.8 (7.6, 9.8) | 12.0 (10.3, 13.5) | 8.5 (7.5, 9.6) |
| Hazard ratio (95% CI)c | 0.73 (0.58, 0.91) | 0.69 (0.56, 0.84) | ||
| p-valued | 0.00306 | 0.00011 | ||
| Progression-free survival (PFS)a | ||||
| Events, n (%) | 197 (82.4%) | 214 (89.9%) | 253 (83.2%) | 269 (88.5%) |
| Median in months (95% CI)b | 2.8 (2.6, 4.0) | 2.9 (2.7, 3.9) | 2.8 (2.6, 3.9) | 2.9 (2.7, 3.4) |
| Hazard ratio (95% CI)c | 0.71 (0.58, 0.86) | 0.75 (0.62, 0.89) | ||
| p-valued | 0.00026 | 0.00048 | ||
| Objective response rate (%)a | ||||
| ORR (95% CI)e | 17.6 (13.0, 23.0) | 6.7 (3.9, 10.7) | 16.4 (12.5,21.1) | 6.3 (3.8, 9.6) |
| Duration of Response (DOR)a | N=42 | N=16 | N=50 | N=19 |
| Median (months)b (95% CI) | 16.4 (12.4, NE) | 6.9 (4.2, 7.7) | 16.4 (12.4, NE) | 6.9 (5.1, 7.7) |
a Median follow-up: 18.2 months. (Data cutoff – Jan 04, 2021)
b Based on Kaplan-Meier estimates.
c Based on stratified proportional hazards model stratified by histology and geographic region.
d One-sided p-value based on stratified proportional hazards model (cemiplimab vs. chemotherapy)
In an updated OS analysis (data cutoff Jan 04, 2022), at a median duration of follow-up of 30.2 months, cemiplimab showed a continued survival benefit compared to chemotherapy (Hazard Ratio (HR): 0.66, 95% CI [0.55, 0.79]) (see Figure 5).
Figure 5. OS in Study 1676 in cervical cancer – Total population (Updated analysis)a:
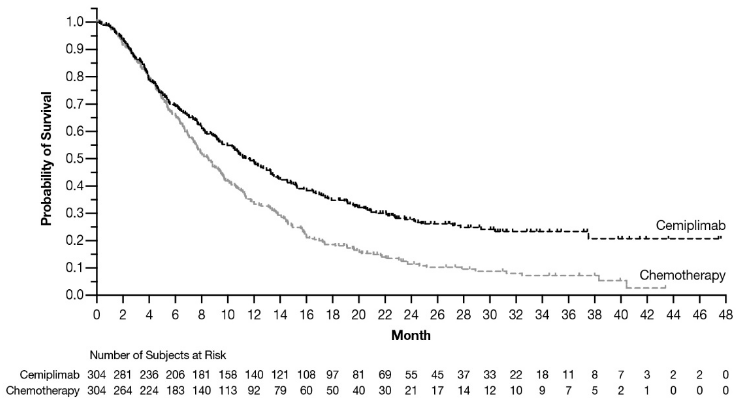
a Based on results from an updated OS analysis which was conducted one year after the primary analysis.
Figure 6. PFS in Study 1676 in cervical cancer – Total population (Primary Analysis):
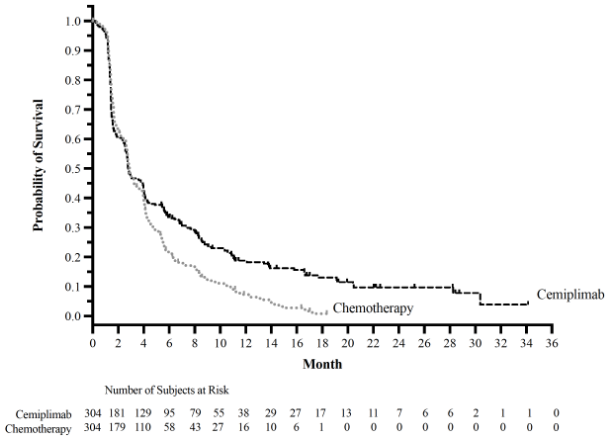
Subgroup analyses
In a subgroup analysis of overall survival by histology based on the updated exploratory OS analysis. the HR for the SCC group was 0.69 (95% CI: 0.56, 0.85) and the HR for the AC group was 0.55 (95% CI: 0.36, 0.81).
An exploratory subgroup analysis was conducted on survival by tumour PD-L1 Tumour Cell (TC) expression status using a clinical trial assay (VENTANA PD-L1 SP263 Assay). Of the 608 enrolled patients, 42% of patients had samples that were tested for PD-L1. Among these samples, 64% were PD-L1 ≥1% and 36% were PD-L1 <1%. At the updated exploratory OS analysis, with median duration of follow-up of 30.2 months, the HR for the PD-L1 ≥1% group was 0.70 (95% CI: 0.48, 1.01) and the HR for the PD-L1 <1% group was 0.85 (95% CI: 0.53, 1.36).
Elderly population
Monotherapy
Of the 1281 patients treated with cemiplimab monotherapy in clinical studies, 52.2% (669/1281) were less than 65 years, 25.9% (332/1281) were 65 to less than 75 years, and 21.9% (280/1281) were 75 years or older.
No overall differences in efficacy were observed between elderly patients and younger patients. There was a trend towards a higher frequency of serious adverse events and discontinuations due to adverse events in patients 65 years and older compared with patients aged less than 65 years treated with cemiplimab monotherapy.
Combination therapy
Of the 312 patients treated with cemiplimab in combination with chemotherapy, 59% (184/312) were less than 65 years, 35.3% (110/312) were 65 to less than 75 years, and 5.8% (18/312) were 75 years or older.
No overall differences in safety or efficacy were observed between elderly patients and younger patients treated with cemiplimab in combination with platinum-based chemotherapy.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with cemiplimab in all subsets of the paediatric population in the treatment of all conditions included in the category of malignant neoplasms, except haematopoietic and lymphoid tissue (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Concentration data from 1063 patients with various solid tumours who received intravenous cemiplimab were combined in a population PK analysis.
At 350 mg Q3W, the mean cemiplimab concentrations at steady-state ranged between a Ctrough of 59 mg/l and a concentration at end of infusion (Cmax) of 171 mg/l. Steady-state exposure is achieved after approximately 4 months of treatment.
Cemiplimab exposure at steady-state in patients with solid tumours is similar at 350 mg Q3W and at 3 mg/kg Q2W.
Absorption
Cemiplimab is administered via the intravenous route and hence is completely bioavailable.
Distribution
Cemiplimab is primarily distributed in the vascular system with a volume of distribution at steady-state (Vss) of 5.9 l. Median Tmax occurs at the end of the 30-minute infusion.
Biotransformation
Specific metabolism studies were not conducted because cemiplimab is a protein. Cemiplimab is expected to degrade to small peptides and individual amino acids.
Elimination
Clearance of cemiplimab is linear at doses of 1 mg/kg to 10 mg/kg every two weeks. Cemiplimab clearance after the first dose is approximately 0.25 l/day. The total clearance appears to decrease by approximately 11% over time, resulting in a steady-state clearance (CLss) of 0.22 l/day; the decrease in CL is not considered clinically relevant. The within dosing interval half-life at steady-state is 22 days.
Linearity/non-linearity
At the dosing regimens of 1 mg/kg to 10 mg/kg every two weeks, pharmacokinetics of cemiplimab were linear and dose proportional, suggesting saturation of the systemic target-mediated pathway.
Special populations
A population PK analysis suggests that the following factors have no clinically significant effect on the exposure of cemiplimab: age, gender, body weight, race, cancer type, albumin level, renal impairment, and mild to moderate hepatic impairment.
Renal impairment
The effect of renal impairment on the exposure of cemiplimab was evaluated by a population PK analysis in patients with mild (CLcr 60 to 89 ml/min; n=396), moderate (CLcr 30 to 59 ml/min; n=166), or severe (CLcr 15 to 29 ml/min; n=7) renal impairment. No clinically important differences in the exposure of cemiplimab were found between patients with renal impairment and patients with normal renal function. Cemiplimab has not been studied in patients with CLcr <21 ml/min (see section 4.2).
Hepatic impairment
The effect of hepatic impairment on the exposure of cemiplimab was evaluated by population PK analysis in patients with mild hepatic impairment (n=22) (total bilirubin [TB] greater than 1.0 to 1.5 times the upper limit of normal [ULN] and any aspartate aminotransferase [AST]) and patients with moderate hepatic impairment (n=3) (total bilirubin >1.5 times ULN up to 3.0 times ULN) and any AST; no clinically important differences in the exposure of cemiplimab were found compared to patients with normal hepatic function. Cemiplimab has not been studied in patients with severe hepatic impairment. There are insufficient data in patients with severe hepatic impairment for dosing recommendations (see section 4.2).
Preclinical safety data
No studies have been performed to test the potential of cemiplimab for carcinogenicity or genotoxicity. Animal reproduction studies have not been conducted with cemiplimab (see section 4.6). As reported in the literature, PD-1/PD-L1 signalling pathway plays a role in sustaining pregnancy by maintaining immunological tolerance and studies have shown that PD-1 receptor blockade results in early termination of pregnancy. The increase of spontaneous abortion and/or resorption in animals with restricted PD-L1 expression (knock-out or anti-PD-1/PD-L1 monoclonal antibodies) has been shown in both mice and monkeys. These animal species have similar maternal-foetal interface to that in humans.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.