LITFULO Hard capsule Ref.[51249] Active ingredients: Ritlecitinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, Janus-associated kinase (JAK) inhibitors
ATC code: L04AF08
Mechanism of action
Ritlecitinib irreversibly and selectively inhibits Janus kinase (JAK) 3 and the tyrosine kinase expressed in hepatocellular carcinoma (TEC) family by blocking the adenosine triphosphate (ATP) binding site. In cellular settings, ritlecitinib specifically inhibits γ-common cytokines (IL-2, IL-4, IL-7, IL-15 and IL-21) signalling through JAK3-dependent common-γ chain receptors. Additionally, ritlecitinib inhibits TEC family of kinases, resulting in reduced cytolytic activity of NK cells and CD8+ T cells.
JAK3 and TEC family mediated signalling pathways are both involved in alopecia areata pathogenesis, although complete pathophysiology is still not understood.
Pharmacodynamic effects
Lymphocyte subsets
In patients with alopecia areata, treatment with ritlecitinib was associated with dose-dependent early decreases in absolute lymphocyte levels, T lymphocytes (CD3) and T lymphocyte subsets (CD4 and CD8). After the initial decrease, the levels partially recovered and remained stable up to 48 weeks. There was no change observed in B lymphocytes (CD19) in any treatment group. There was a dose-dependent early decrease in NK cells (CD16/56) which remained stable at the lower level up to Week 48.
Immunoglobulins
In patients with alopecia areata, treatment with ritlecitinib was not associated with clinically meaningful changes in Immunoglobulin (Ig)G, IgM or IgA up to Week 48, indicating a lack of systemic humoral immunosuppression.
Clinical efficacy and safety
The efficacy and safety of ritlecitinib was evaluated in a pivotal, randomised, double-blind, placebo-controlled study (study AA-I) in alopecia areata patients 12 years of age and older with ≥50% scalp hair loss, including alopecia totalis and alopecia universalis. The dose-response of ritlecitinib was also evaluated in this study. The study treatment period consisted of a placebo-controlled 24-week period and a 24-week extension period. Study AA-I evaluated a total of 718 patients who were randomised to one of the following treatment regimens for 48 weeks: 1) 200 mg once daily for 4 weeks followed by 50 mg once daily for 44 weeks; 2) 200 mg once daily for 4 weeks followed by 30 mg once daily for 44 weeks; 3) 50 mg once daily for 48 weeks; 4) 30 mg once daily for 48 weeks; 5) 10 mg once daily for 48 weeks; 6) placebo for 24 weeks followed by 200 mg once daily for 4 weeks and 50 mg once daily for 20 weeks; or 7) placebo for 24 weeks followed by 50 mg for 24 weeks.
This study assessed as primary outcome the proportion of subjects who achieved a SALT (Severity of Alopecia Tool) score of ≤10 (90% or more scalp hair coverage) at Week 24. Additionally, this study assessed as key secondary outcome the Patient’s Global Impression of Change (PGI-C) response at Week 24 and also assessed as secondary outcomes SALT score of ≤20 (80% or more scalp hair coverage) at Week 24 and improvements in regrowth of eyebrows and/or eyelashes at Week 24.
Baseline characteristics
Male or female patients 12 years of age and older, were assessed in study AA-I. All patients had alopecia areata with ≥50% scalp hair loss (SALT [Severity of Alopecia Tool] score ≥50) without evidence of terminal hair regrowth within the previous 6 months and with the current episode of scalp hair loss ≤10 years and no other known cause of hair loss (e.g., androgenetic alopecia).
Across all treatment groups 62.1% were female, 68.0% were White, 25.9% were Asian, and 3.8% were Black or African American. The mean age of patients was 33.7 years and the majority (85.4%) were adults (≥18 years of age). A total of 105 (14.6%) patients 12 to <18 years of age and 20 (2.8%) patients 65 years of age and older were enrolled. The mean (SD) baseline absolute SALT score ranged from 88.3 (16.87) to 93.0 (11.50) across treatment groups; among patients without alopecia totalis/alopecia universalis at baseline, the mean SALT score ranged from 78.3 to 87.0. The majority of patients had abnormal eyebrows (83.0%) and eyelashes (74.7%) at baseline across treatment groups. The median duration since alopecia areata diagnosis was 6.9 years and the median duration of the current alopecia areata episode was 2.5 years. Randomisation was stratified by alopecia totalis/alopecia universalis status with 46% of patients classified as alopecia totalis/alopecia universalis based upon a baseline SALT score of 100.
Clinical response
A significantly greater proportion of patients achieved SALT ≤10 response with ritlecitinib 50 mg compared to placebo at Week 24 (Table 3). The SALT ≤10 response rate for ritlecitinib 50 mg increased further at Week 48 (Figure 1).
A significantly greater proportion of patients achieved Patient’s Global Impression of Change (PGI-C) response with ritlecitinib 50 mg compared to placebo at Week 24 (Table 3) with response rates continuing to increase through Week 48 (Figure 1).
A significantly greater proportion of patients achieved a SALT ≤20 response with ritlecitinib 50 mg compared to placebo at Week 24 (Table 3). The SALT ≤20 response rate increased further at Week 48.
Improvements in regrowth of eyebrows and/or eyelashes were seen at Week 24 (Table 3) with ritlecitinib 50 mg among patients with abnormal eyebrows and/or eyelashes at baseline with further increases seen at Week 48.
Treatment effects at Week 24 in subgroups (age, gender, race, region, weight, duration of disease since diagnosis, duration of current episode, prior pharmacologic treatment) were consistent with the results in the overall study population. Treatment effects at Week 24 in the alopecia totalis/alopecia universalis subgroup were lower compared to the non-alopecia totalis/non-alopecia universalis subgroup. Treatment effects at Week 24 in adolescents 12 to less than 18 years of age were consistent with the results in the overall study population.
Table 3. Efficacy results of ritlecitinib at week 24:
| Endpoint | Ritlecitinib 50 mg once daily (N=130) % Responders | Placebo (N=131) % Responders | Difference from placebo (95% CI) |
|---|---|---|---|
| SALT ≤10 responsea,b | 13.4 | 1.5 | 11.9 (5.4, 18.3) |
| PGI-C responseb,c | 49.2 | 9.2 | 40.0 (28.9, 51.1) |
| SALT ≤20 responsed,e | 23.0 | 1.6 | 21.4 (13.4, 29.5) |
| EBA responsef | 29.0 | 4.7 | 24.3 (14.8, 34.5) |
| ELA responseg | 28.9 | 5.2 | 23.7 (13.6, 34.5) |
Figure 1. SALT ≤10 and PGI-C response through Week 48:
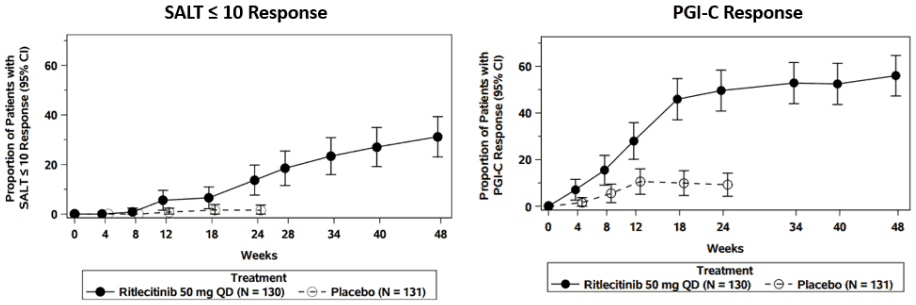
Abbreviations: CI = confidence interval; N = total number of patients; PGI-C = Patient Global Impression of Change; QD = once daily; SALT = Severity of Alopecia Tool
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with ritlecitinib in one or more subsets of the paediatric population in the treatment of alopecia areata (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
The absolute oral bioavailability of ritlecitinib is about 64%. Based on oral and intravenous administration of the labelled active substance, the relative urinary recovery (oral/intravenous) of labelled compounds was about 89%, indicating a high fraction absorbed (fa). Peak plasma concentrations are reached within 1 hour following multiple oral doses. Food does not have a clinically significant impact on the extent of ritlecitinib absorption, as a high-fat meal decreased the ritlecitinib Cmax by ~32% and increased AUCinf by ~11%. In placebo-controlled studies, ritlecitinib was administered without regard to meals (see section 4.2).
In vitro, ritlecitinib is a substrate of P-glycoprotein (P-gp) and BCRP. However, as ritlecitinib has a high fraction absorbed (fa) with both Cmax and AUC increases in a dose proportional manner (20–200 mg single dose range), P-gp and BCRP are not expected to have a meaningful impact on the absorption of ritlecitinib.
Distribution
After intravenous administration, the volume of distribution of ritlecitinib is about 74 L. Approximately 14% of circulating ritlecitinib is bound to plasma proteins, primarily albumin. The blood/plasma distribution ratio of ritlecitinib is 1.62. Ritlecitinib is a covalent inhibitor that has been shown to bind to off-target proteins such as MAP2K7, DOCK10, albumin, CYP1A2, CYP3A, UGT1A1, and UGT1A4, some of which may have clinical relevance in drug interactions (see section 4.5).
Biotransformation
The metabolism of ritlecitinib is mediated by multiple isoforms of Glutathione S-transferase (GST: cytosolic GST A1/3, M1/3/5, P1, S1, T2, Z1, and microsomal Membrane Associated Proteins involved in Eicosanoid and Glutathione metabolism [MAPEG]1/2/3) and CYP enzymes (CYP3A, CYP2C8, CYP1A2, and CYP2C9), with no single clearance route contributing more than 25%. Hence, medicinal products inhibiting a selective metabolic pathway are unlikely to impact the systemic exposures of ritlecitinib. Specific inhibitors of transporters are unlikely to result in clinically relevant changes in the bioavailability of ritlecitinib.
In a human radiolabeled study, ritlecitinib was the most prevalent circulating species (30.4% of circulating radioactivity) after oral administration, with a major cysteine conjugate metabolite M2 (16.5%), which is pharmacologically inactive.
Elimination
Ritlecitinib is eliminated primarily by metabolic clearance mechanisms, with approximately 4% of the dose excreted as unchanged active substance in urine. Approximately 66% of radiolabeled ritlecitinib dose is excreted in the urine and 20% in the faeces. Following multiple oral doses, steady state was reached approximately by Day 4 due to non-stationary PK. The steady state PK parameters of AUCtau and Cmax appeared to increase in an approximately dose-proportional manner up to 200 mg with the mean terminal half-life ranging from 1.3 to 2.3 hours.
Special populations
Body weight, gender, genotype, race and age
Body weight, gender, GST P1, M1, and T1 genotype, race and age did not have a clinically meaningful effect on ritlecitinib exposure.
Adolescents (≥12 to <18 years)
Based on population PK analysis, there was no clinically relevant difference in ritlecitinib exposures in adolescent patients compared to adults.
Paediatric (<12 years)
The PK of ritlecitinib in children under 12 years of age have not yet been established.
Renal impairment
The AUC24 and Cmax in patients with severe renal impairment (estimated glomerular filtration rate [eGFR] <30 mL/min) was about 55% and 44% higher, respectively, compared with matched participants with normal renal functions. This was confirmed by popPK analysis. These differences are not considered clinically significant. Ritlecitinib was not studied in patients with mild (eGFR 60 to <90 mL/min) or moderate (eGFR 30 to <60 mL/min) renal impairment. However, based on the results obtained in patients with severe renal impairment, a clinically significant increase in ritlecitinib exposure is not expected in these patients. The eGFR and classification of renal function status of participants was done using the Modification of Diet in Renal Disease (MDRD) formula.
Based on the above considerations, no dose adjustment is required in patients with mild, moderate or severe renal impairment. Ritlecitinib has not been studied in patients with ESRD or in renal transplant recipients (see section 4.2).
Hepatic impairment
Patients with moderate (Child Pugh B) hepatic impairment had an 18.5% increase in ritlecitinib AUC24 compared to participants with normal hepatic function. Ritlecitinib was not studied in patients with mild (Child Pugh A) hepatic impairment. However, based on the results obtained in patients with moderate hepatic impairment, a clinically significant increase in ritlecitinib exposure is not expected in these patients. No dose adjustment is required in patients with mild or moderate hepatic impairment (see section 4.2). Ritlecitinib has not been studied in patients with severe (Child Pugh C) hepatic impairment (see section 4.3).
5.3. Preclinical safety data
General toxicity
Decreased lymphocyte counts and decreased lymphoid cellularity of organs and tissues of the immune and haematolymphopoietic systems were observed in nonclinical toxicity studies and were attributed to the pharmacological properties (JAK3/TEC inhibition) of ritlecitinib.
Chronic administration of ritlecitinib to Beagle dogs led to the occurrence of axonal dystrophy at systemic exposures of at least 7.4-times the expected exposure in patients treated with 50 mg per day (based on unbound AUC24). Axonal dystrophy is presumably related to binding to off-target neuronal proteins. It is not known if axonal dystrophy occurred in dogs at lower systemic exposures. At a systemic exposure that was 33-times above the expected exposure in patients treated with 50 mg per day (based on unbound AUC24), axonal dystrophy was associated with neurological hearing loss. While these findings proved to reverse after dosing cessation of ritlecitinib in dogs, a risk to patients at a chronic dosing regimen cannot be fully excluded (see section 4.4).
Genotoxicity
Ritlecitinib was not mutagenic in the bacterial mutagenicity assay (Ames assay). Ritlecitinib is not aneugenic or clastogenic at exposures equal to 130 times the MRHD on an unbound AUC basis based on the results of the in vivo rat bone marrow micronucleus assay.
Carcinogenicity
No evidence of tumorigenicity was observed in the 6-month Tg.ras H2 mice administered ritlecitinib at exposures equal to 11 times the MRHD on an unbound AUC basis. In a 2-year rat carcinogenicity study, a higher incidence of benign thymomas in female rats and benign thyroid follicular adenomas in male rats was noted following ritlecitinib administration at exposures equal to 29 times the MRHD on an unbound AUC basis. At this ritlecitinib exposure, a higher incidence of malignant thymomas in female rats cannot be excluded. No ritlecitinib-related thymomas or thyroid follicular adenomas were observed at exposures equal to 6.3 times the MRHD on an unbound AUC basis.
Reproductive and developmental toxicity
Ritlecitinib had no effects on female rat fertility at exposures equal to 55 times the MRHD on an unbound AUC basis. Effects on male rat fertility were noted (higher preimplantation loss resulting in lower number of implantation sites and corresponding lower litter size in naïve females mated with ritlecitinib dosed males) at exposure equal to 55 times the MRHD on an unbound AUC basis. No effects on male fertility were noted at exposures equal to 14 times the MRHD on an unbound AUC basis. No effects on spermatogenesis (sperm counts, sperm production rate, motility, and morphology) were noted at any dose in the rat fertility study.
In an embryo-foetal development study in pregnant rats, oral administration of ritlecitinib from gestation days 6 to 17 resulted in foetal skeletal malformations and variations and lower foetal body weights at exposures greater than or equal to 49 times the unbound AUC at the MRHD (see section 4.3). There were no effects on embryo-foetal development at exposures equal to 16 times the unbound AUC at the MRHD.
In an embryo-foetal development study in pregnant rabbits, oral administration of ritlecitinib from gestation days 7 to 19 resulted in lower mean foetal body weights and higher incidences of visceral malformations, skeletal malformations, and skeletal variations at exposures equal to 55 times the unbound AUC at the MRHD (see section 4.3). There were no effects on embryo-foetal development at exposures equal to 12 times the unbound AUC at the MRHD.
In a rat pre- and postnatal development study, oral administration of ritlecitinib from gestation day 6 through lactation day 20 resulted in developmental toxicity that included lower postnatal survival, lower offspring body weights, and secondary developmental delays at exposure equal to 41 times the unbound AUC at the MRHD (see section 4.3). Bred females in the F1 generation exhibited lower mean numbers of corpora lutea at exposures equal to 41 times the unbound AUC at the MRHD. There were no effects on pre- and postnatal development at exposures equal to 14 times the unbound AUC at the MRHD.
In a juvenile rat toxicity study, oral administration of ritlecitinib from postnatal day 10 to 60 (comparable to infant through adolescence human age) was not associated with effects on the nervous or skeletal systems.
Lactation
Following administration of ritlecitinib to lactating rats, concentrations of ritlecitinib in milk over time were higher than those in plasma, where the mean milk to plasma AUC ratio was determined to be 2.2 (see section 4.3).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.