LIVMARLI Oral solution Ref.[50565] Active ingredients: Maralixibat
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Mirum Pharmaceuticals International B.V., Kingsfordweg 151, 1043 GR Amsterdam, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Bile and liver therapy, other drugs for bile therapy
ATC code: A05AX04
Mechanism of action
Maralixibat is a minimally absorbed, reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).
Maralixibat acts locally in the distal ileum to decrease the reuptake of bile acids and increase the clearance of bile acids through the colon, reducing the concentration of bile acids in the serum.
Clinical efficacy and safety
The efficacy of maralixibat in ALGS patients was assessed in a 48-week trial which included an 18-week open-label active substance run-in period, a 4-week double-blind randomised withdrawal period and a long-term, open-label extension period.
Thirty-one ALGS paediatric patients with cholestasis and pruritus were enrolled, with 90.3% of patients receiving at least one medication to treat pruritus at trial entry (74.2% and 80.6% of patients receiving rifampicin and ursodeoxycholic acid, respectively). Concomitant use of these medications was allowed during the trial, but dose adjustments were prohibited during the first 22 weeks. All patients had ALGS due to JAGGED1 mutation.
Exclusion criteria included surgical interruption of the enterohepatic circulation, history or presence of any condition known to interfere with the absorption, distribution, metabolism or excretion of drugs, including bile salt metabolism in the intestine, and chronic diarrhoea requiring intravenous fluid or nutritional intervention.
After an initial 5-week dose-escalation period, patients were administered open-label treatment with maralixibat 380 mcg/kg once daily for 13 weeks; two patients discontinued treatment during this first 18 weeks of open-label run-in treatment. The 29 patients who completed the open-label run-in phase were then randomised to either continue treatment with maralixibat or receive matching placebo (n=16 placebo, n=13 maralixibat) during the 4-week double-blind randomised withdrawal period at weeks 19-22. All 29 patients completed the blinded randomised withdrawal period; subsequently, all patients received open-label maralixibat at 380 mcg/kg once daily dose for up to 48 weeks. Patients who were switched from placebo went through a dose escalation schedule similar to the initial escalation.
Randomised patients had a median age of 5 years (range: 1 to 15 years) and 66% were male. The baseline mean (standard deviation [SD]) of liver test parameters were as follows: serum bile acid (sBA) levels 280 (213) µmol/L, aspartate aminotransferase (AST) 158 (68) U/L, alanine transaminase (ALT) 179 (112) U/L, gamma glutamyl transferase (GGT) 498 (399) U/L, and total bilirubin (TB) 5.6 (5.4) mg/dL.
Serum bile acids (sBA)
A statistically significant mean (SD) reduction in sBA versus baseline of 88 (120) and 96 (166.6) μmol/L was observed at week 18 and week 48 when patients were administered maralixibat. At the end of the placebo-controlled period, a statistically significant least squares mean (SE) difference was demonstrated between maralixibat and placebo in change in sBA from week 18 to week 22 (-114 [48.0] µmol/L; p=0.025). When the placebo group resumed treatment with maralixibat at the end of the withdrawal period, sBA reduced to levels previously observed with maralixibat treatment (see Figure 1).
Figure 1. Mean (± SE) change from baseline sBA, through week 48, all patients:

Pruritus
Pruritus severity was evaluated in the overall population (n=31), measured by Itch Reported Outcome Observer (ItchRO[Obs]) score. The ItchRO score is a validated 0-4 scale completed by caregivers (0=none to 4=very severe), where changes ≥1.0 have been shown to be clinically meaningful. Changes in pruritus severity between participants treated with maralixibat and those treated with placebo during the randomised withdrawal period and changes from baseline to week 18 and to week 48 were measured. The mean ItchRO(Obs) score at baseline was 2.9.
Patients administered maralixibat demonstrated a clinically meaningful change and statistically significant reductions of ItchRO(Obs) of -1.7 and -1.6 points from baseline at week 18 and week 48, respectively.
During the placebo-controlled randomised withdrawal period, patients administered maralixibat maintained pruritus reduction, whereas those in the placebo group returned to baseline pruritus scores. The difference between maralixibat and placebo in least squares mean (SE) change in pruritus from week 18 to week 22 (-1.5 [0.3]; 95% CI: -2.1 to -0.8; p<0.0001; see Figure 2) was statistically significant. After resuming maralixibat, patients from the placebo group regained improvement in pruritus by week 28. Patients administered maralixibat demonstrated sustained pruritus reduction up to 48 weeks.
Figure 2. ItchRO(Obs) weekly average morning severity score change from baseline by randomised treatment group over time, through week 48, all patients:

Improvements of variable degree in cholesterol and xanthoma severity were observed during treatment with maralixibat.
The mechanism of action of maralixibat to prevent reuptake of bile acids is expected to be similar across all age groups. Evidence of efficacy in patients younger than 12 months of age with ALGS is limited. In an open-label, single-arm study in 8 patients of 2 to 10 months of age with ALGS change in pruritus as assessed with Clinician Scratch Scale (where 0 = none and 4 = cutaneous mutilation, haemorrhage and scarring evident) at week 13 was mean (SD; median; range) -0.2 (1.91; -1.0; -3.0 to 3.0) and in sBA mean (SD; median; range) -88.91 µmol/L (113.348; -53.65; -306.1 to 14.4). Two patients experienced improvement in both pruritus and sBA.
Clinical efficacy in PFIC
The efficacy of maralixibat was assessed in a 26-week randomized, double-blind placebo-controlled trial (MRX-502). Ninety-three patients with diagnosis of PFIC based on documentation of intrahepatic cholestasis with persistent pruritus, abnormal tests for liver function and/or evidence of progressive liver disease aged >12 months and <18 years were included. Patients underwent genotyping for confirmation of PFIC type. Persistent pruritus was defined as > 6 months with average pruritus score on ItchRO[Obs] equal or greater than 1.5 in the 4 weeks prior to baseline.
Patients with decompensated cirrhosis, history or presence of any condition known to interfere with the absorption, distribution, metabolism or excretion of drugs, including bile salt metabolism in the intestine, and chronic diarrhoea requiring intravenous fluid or nutritional intervention were excluded.
Patients were randomized 1:1 to receive maralixibat 570 mcg/kg (n=47) or placebo orally (n=46) twice daily for 26 weeks with an initial 4–6-week dose escalation period, starting with 142 mcg/kg twice daily. The 26-week study period was completed by 92.5% of patients (44/47 maralixibat and 42/46 placebo), with 7 discontinuing from the study (4 withdrawal of consent, 1 AE for mild diarrhoea, 1 liver transplantation, and 1 disease progression). Patients completing the pivotal trial were eligible to enrol in an open-label extension trial (MRX-503).
Efficacy endpoints for the pivotal trial included changes in pruritus severity, serum bile acid levels, liver function tests and growth.
Efficacy endpoints were evaluated in patients with genetic testing results consistent with biallelic PFIC-causing variants (n=64): ABCB11/BSEP (PFIC2) n=31; ATP8B1/FIC1 (PFIC1) n=13; ABCB4/MDR3 (PFIC3) n=9; TJP2 (PFIC4) n=7; MYO5B (PFIC 6) n=4. There were more females (53.1%) and the mean age was 4.6 years with a range of 1 to 15 years. Most patients were on stable ursodeoxycholic acid (89.1%) or rifampicin (51.6%) therapy at baseline. The baseline mean (standard deviation [SD]) of liver test parameters were as follows: serum bile acid levels 263 (143) μmol/L, AST 113 (82) U/L, ALT 107 (87) U/L, and TB 69.8 (70.1) μmol/L, DB 50.6 (52.4) μmol/L. The mean (SD) of the average baseline morning ItchRO[Obs] pruritus severity score was 2.8 (0.87). There were no meaningful differences observed between treatment groups across baseline characteristics or disease parameters.
Serum bile acids (sBA)
The mean change in total serum bile acid level between maralixibat and placebo treatment groups from baseline to average of weeks 18, 22, and 26 was statistically significant with a LS mean change from placebo of -160 μmol/L (95% CI: -220.8, -100.0) (Figure 3).
Figure 3. Observed average serum bile acids levels over time in PFIC 1, 2, 3, 4, and 6 (Study MRX-502):
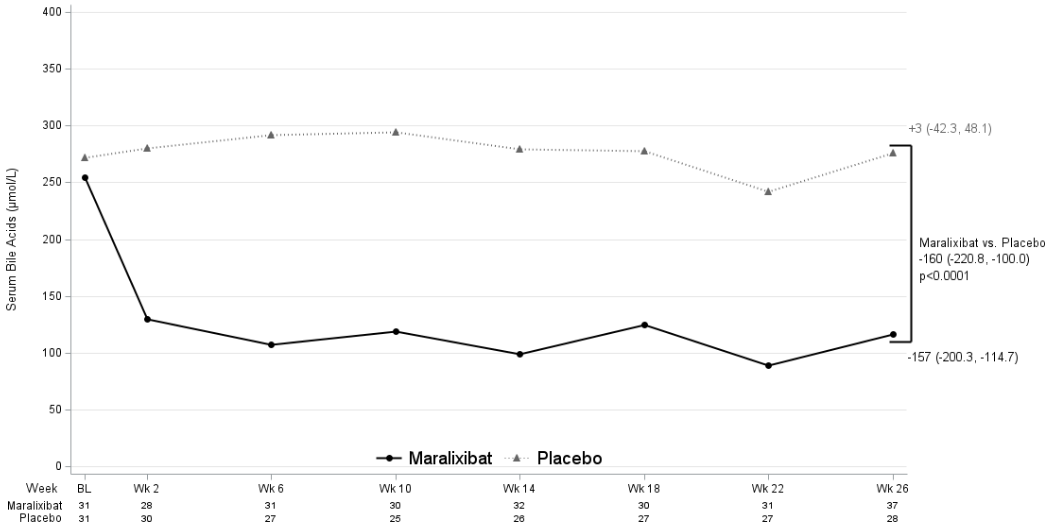
The percentage of serum bile acid responders was 45.5% for maralixibat and 6.5% for placebo participants, difference (95% CI): 39.0% (16.5%, 58.2%). Serum bile acid responders were defined as a participant having an average sBA level of <102 μmol/L (applies only if baseline sBA level was ≥102 μmol/L) OR ≥75% average reduction from baseline. For the purpose of determining response, the average sBA value from weeks 18, 22, and 26 values were used.
Pruritus
Maralixibat demonstrated difference between maralixibat and placebo treatment groups for the average change in morning ItchRO(Obs) severity score between baseline and weeks 15–26, with a LS mean change from placebo -1.200 (95% CI: -1.727, -0.674; Figure 4).
Figure 4. Observed weekly average of the morning daily pruritus score over time in PFIC 1, 2, 3, 4, and 6 (Study MRX-502):
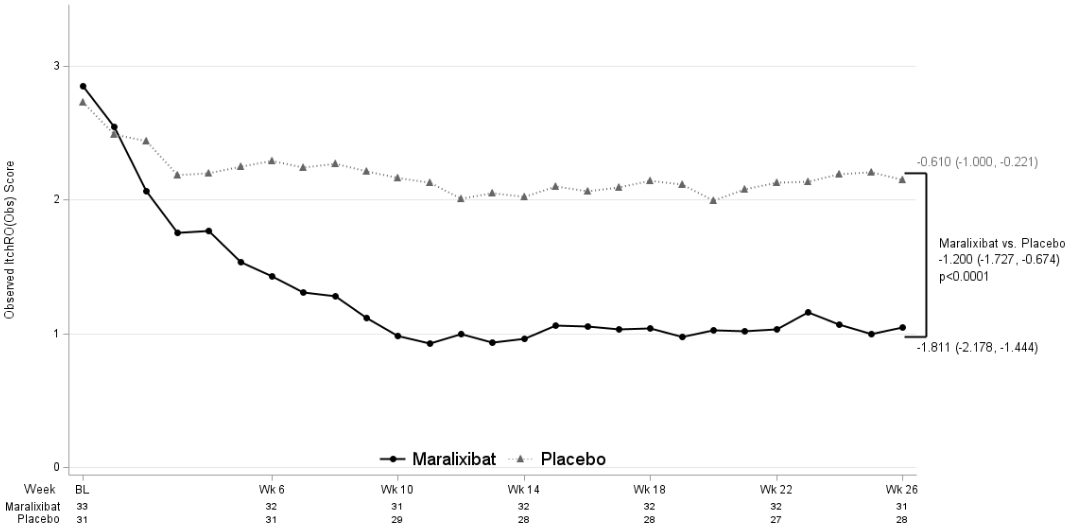
BL=Baseline; Wk=Week. Observed values are displayed. Statistics shown are averages of time periods weeks 15-18, 19-22, and 23-26 using an equally weighted average of the 3 individual visit-specific estimates obtained from a mixed model for repeated measures (MMRM) with change from baseline as the dependent variable and fixed categorical effects of treatment group, PFIC type, analysis visit and treatment-by-visit interaction as well as the continuous fixed covariates of baseline score and baseline score-by-visit interaction. The least-squares mean estimate and 95% confidence interval are presented.
Table 4 presents the results of the comparison of the ItchRO(Obs) results between maralixibat and placebo.
Table 4. Proportion of pruritus responders (Study MRX-502):
| Responder Type Category | Maralixibat (n=33) | Placebo (n=31) |
|---|---|---|
| ItchRO(Obs) responders; average score ≤1 OR change from baseline of ≤-1.0 | ||
| Responder (%) | 63.6 | 25.8 |
| p-value vs. placebo difference (95% CI) | 0.0023 | 37.8 (11.3, 59.4) |
p-values comparing maralixibat to placebo treatment groups are calculated using a Barnard’s exact test.
Exact 95% confidence intervals are based on a score statistic.
Exploratory analyses showed more pronounced reduction (improvement) in the mean sleep disturbance scores in the maralixibat treatment group compared with placebo. Exploratory analyses showed improvements in bilirubin during treatment with maralixibat (Table 5). Abnormal total bilirubin levels at baseline normalised by week 26 in 40% (10/25) of patients on maralixibat vs. 0% (0/18) on placebo. More pronounced increase (improvement) in weight z-score was observed in the maralixibat treatment group compared with placebo (LS mean change from placebo of 0.227 (95% CI: 0.012, 0.442; Table 5)).
Table 5. Liver function tests and growth parameters for maralixibat vs. placebo over the 26-week treatment period in participants with PFIC in the pivotal trial (MRX-502 exploratory analyses):
| Efficacy endpoint | Placebo (n=31) | Maralixibat (n=33) |
|---|---|---|
| Alanine aminotransferase (U/L) | ||
| Baseline (mean [SE]) | 127.3 (18.68) | 87.8 (10.77) |
| LS mean change from BL [SE] to weeks 18-26 | -7.0 (11.13) | 9.7 (10.36) |
| LS mean difference vs. placebo (95% CI); | 16.6 (-13.31, 46.60) | |
| Aspartate aminotransferase (U/L) | ||
| Baseline (mean [SE]) | 129.8 (18.12) | 96.9 (9.57) |
| LS mean change from BL [SE] to weeks 18-26 | -0.4 (14.91) | 13.6 (14.05) |
| LS mean difference vs. placebo (95% CI); | 14.1 (-26.57, 54.69) | |
| Total bilirubin (μmol/L) | ||
| Baseline (mean [SE]) | 69.1 (13.69) | 70.4 (11.32) |
| LS mean change from BL [SE] to weeks 18-26 | 15.9 (12.37) | -18.3 (11.65) |
| LS mean difference vs. placebo (95% CI); | -34.3 (-68.06, -0.46) | |
| Direct bilirubin (μmol/L) | ||
| Baseline (mean [SE]) | 50.2 (10.28) | 50.9 (8.40) |
| LS mean change from BL [SE] to weeks 18-26 | 13.5 (9.52) | -12.9 (8.97) |
| LS mean difference vs. placebo (95% CI); | -26.4 (-52.46, -0.26) | |
| 3< Height z-score | ||
| Baseline (mean [SE]) | -2.06 (0.27) | -2.08 (0.23) |
| LS mean change from BL [SE] to weeks 18-26 | -0.13 (0.09) | 0.08 (0.09) |
| LS mean difference vs. placebo (95% CI); | 0.21 (-0.04, 0.5) | |
| 3< Weight z-score | ||
| Baseline (mean [SE]) | -1.28 (0.24) | -1.75 (0.23) |
| LS mean change from BL [SE] to weeks 18-26 | 0.12 (0.08) | 0.35 (0.07) |
| LS mean difference vs. placebo (95% CI); | 0.23 (0.01, 0.4) | |
SE = standard error; LS = least-squares; CI=confidence interval; BL = baseline. Baseline values are observed values. LS mean values are averages of weeks 18, 22, and 26 using an equally weighted average of the 3 individual visit-specific estimates obtained from a mixed model for repeated measures (MMRM) with change from baseline as the dependent variable and fixed categorical effects of treatment group, PFIC type, analysis visit and treatment-by-visit interaction as well as the continuous fixed covariates of baseline score and baseline score-by-visit interaction
Of the 64 patients from the pivotal trial (MRX-502) with genetic testing results consistent with biallelic PFIC-causing variants, 57 were included in an interim analysis from the ongoing open-label extension trial (MRX-503). Their median treatment duration with maralixibat was 47.3 weeks (range: 4.1 weeks – 119.4 weeks). Maralixibat showed maintenance of treatment effect on serum bile acid and bilirubin levels as well as pruritus. Height and weight z-scores were further improved.
In an open-label, single-arm safety study (MRX-801) in 10 patients of 1 to 11 months of age with PFIC (no requirement for active pruritus), decrease at week 13 in sBA, total bilirubin and direct bilirubin was observed in some patients. Two patients also experienced improvement in pruritus.
Exceptional circumstances
This medicinal product has been authorised under ‘exceptional circumstances’. This means that due to the rarity of the disease it has not been possible to obtain complete information on this medicinalproduct. The European Medicines Agency will review any new information which may become available every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Absorption
The target of maralixibat is in the lumen of the small intestine, such that plasma levels of maralixibat are not required and not relevant to its efficacy. Maralixibat is minimally absorbed, and plasma concentrations are often below the limit of detection (0.25 ng/mL) after single or multiple doses at therapeutic dose levels. The absolute bioavailability is estimated to be <1%.
Effect of food
Maralixibat absorption is relatively higher when administered in the fasted state, and no dose adjustment for food effects is necessary. Maralixibat can be taken before (up to 30 minutes) or with a meal (see section 4.2).
Distribution
Maralixibat shows high binding (91%) to human plasma in vitro.
In a clinical ADME trial dosing [14C] maralixibat, circulating radioactivity was below the limit of detection at all time points. There is no apparent accumulation of maralixibat.
Biotransformation
No metabolites have been detected in plasma, and maralixibat also undergoes minimal metabolism in the gastrointestinal tract.
Elimination
Maralixibat is primarily eliminated in the faeces as unmetabolised parent compound, with 0.066% of the administered dose excreted in the urine.
Special populations
No clinically significant differences in the pharmacokinetics of maralixibat were observed based on age, sex, or race.
Hepatic impairment
Clinical studies of maralixibat included ALGS and PFIC patients with some level of liver impairment. The majority of patients presented with some degree of hepatic impairment according to the NCI-ODWG classification due to the disease. Whether this classification is, however, appropriate in cholestatic disease to predict the influence on PK of the compound is currently unclear. Maralixibat is minimally absorbed, and animal data indicate that the very low plasma levels are due to low absorption and not a first pass effect in the liver, and plasma levels of maralixibat were not increased in patients with liver impairment according to the NCI-ODWG. However, the PK of maralixibat have not been systematically investigated in patients classified according to the Child-Pugh classification (patients with cirrhosis and signs of decompensation).
Renal impairment
The pharmacokinetics of maralixibat were not studied in patients with impaired renal function, including those with ESRD or those on haemodialysis. However, renal impairment is not expected to impact maralixibat PK due to the low systemic exposure and lack of urinary excretion.
5.3. Preclinical safety data
Non-clinical data reveal no specific hazard for humans based on studies of safety pharmacology, secondary pharmacology, repeated-dose toxicity, genotoxicity, carcinogenicity, fertility, toxicity to reproduction and development, and juvenile animal toxicity.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.