LORVIQUA Film-coated tablet Ref.[7603] Active ingredients: Lorlatinib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
Pharmacodynamic properties
Pharmacotherapeutic group: anti-neoplastic agents, protein kinase inhibitors
ATC code: L01ED05
Mechanism of action
Lorlatinib is a selective, adenosine triphosphate (ATP)-competitive inhibitor of ALK and c-ros oncogene 1 (ROS1) tyrosine kinases.
In non-clinical studies, lorlatinib inhibited catalytic activities of non-mutated ALK and clinically relevant ALK mutant kinases in recombinant enzyme and cell-based assays. Lorlatinib demonstrated 15 marked antitumour activity in mice bearing tumour xenografts that express echinoderm microtubule-associated protein-like 4 (EML4) fusions with ALK variant 1 (v1), including ALK mutations L1196M, G1269A, G1202R, and I1171T. Two of these ALK mutants, G1202R and I1171T, are known to confer resistance to alectinib, brigatinib, ceritinib, and crizotinib. Lorlatinib was also capable of penetrating the blood-brain barrier. Lorlatinib demonstrated activity in mice bearing orthotopic EML4-ALK or EML4-ALKL1196M brain tumour implants.
Clinical efficacy
Previously untreated ALK-positive advanced NSCLC (CROWN Study)
The efficacy of lorlatinib for the treatment of patients with ALK-positive NSCLC who had not received prior systemic therapy for metastatic disease was established in an open-label, randomised, active-controlled, multicentre Study B7461006 (CROWN study). Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx assay. Neurologically stable patients with treated or untreated asymptomatic CNS metastases, including leptomeningeal metastases, were eligible. Patients were required to have finished radiation therapy, including stereotactic or partial brain irradiation within 2 weeks prior to randomisation; whole brain irradiation within 4 weeks prior to randomisation.
Patients were randomised 1:1 to receive lorlatinib 100 mg orally once daily or crizotinib 250 mg orally twice daily. Randomisation was stratified by ethnic origin (Asian vs. non-Asian) and the presence or absence of CNS metastases at baseline. Treatment on both arms was continued until disease progression or unacceptable toxicity. The major efficacy outcome measure was progression-free survival (PFS) as determined by Blinded Independent Central Review (BICR) according to Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1 (v1.1). Additional efficacy outcome measures were overall survival (OS), PFS by investigator assessment, PFS2 and tumour assessment related data by BICR, including objective response rate (ORR), duration of response (DOR) and time to intracranial progression (IC-TTP). In patients with CNS metastases at baseline, additional outcome measures were intracranial objective response rate (IC-ORR) and intracranial duration of response (IC-DOR) all by BICR.
A total of 296 patients were randomised to lorlatinib (n=149) or crizotinib (n=147). The demographic characteristics of the overall study population were: median age 59 years (range: 26 to 90 years), age ≥65 years (35%), 59% female, 49% White, 44% Asian and 0.3% Black. The majority of patients had adenocarcinoma (95%) and never smoked (59%). Central nervous system metastases as determined by BICR neuroradiologists were present in 26% (n=78) of patients: of these, 30 patients had measurable CNS lesions.
Results from the CROWN study are summarised in Table 3. At the data cutoff point, OS and PFS2 data were not mature.
Table 3. Overall efficacy results in CROWN study:
| Efficacy parameter | Lorlatinib N=149 | Crizotinib N=147 |
|---|---|---|
| Median duration of follow-up, months (95% CI)a | 18 (16, 20) | 15 (13, 18) |
| Progression-free survival by BICR | ||
| Number of patients with event, n (%) | 41 (28%) | 86 (59%) |
| Progressive disease, n (%) | 32 (22%) | 82 (56%) |
| Death, n (%) | 9 (6%) | 4 (3%) |
| Median, months (95% CI)a | NE (NE, NE) | 9 (8, 11) |
| Hazard ratio (95% CI)b | 0.28 (0.19, 0.41) | |
| p-value* | <0.0001 | |
| Overall survival | ||
| Number of patients with event, n (%) | 23 (15%) | 28 (19%) |
| Median, months (95% CI)a | NE (NE, NE) | NE (NE, NE) |
| Hazard ratio (95% CI)b | 0.72 (0.41, 1.25) | |
| Progression-free survival by INV | ||
| Number of patients with event, n (%) | 40 (27%) | 104 (71%) |
| Progressive disease, n (%) | 34 (23%) | 99 (67%) |
| Death, n (%) | 6 (4%) | 5 (3%) |
| Median, months (95% CI)a | NE (NE, NE) | 9 (7, 11) |
| Hazard ratio (95% CI)b | 0.21 (0.14, 0.31) | |
| p-value* | <0.0001 | |
| Overall response by BICR | ||
| Overall response rate, n (%) | 113 (76%) | 85 (58%) |
| (95% CI)c | (68, 83) | (49, 66) |
| Time to intracranial progression | ||
| Median, months (95% CI)a | NE (NE, NE) | 16.6 (11, NE) |
| Hazard ratio (95% CI)b | 0.07 (0.03, 0.17) | |
| Duration of response | ||
| Number of responders | 113 | 85 |
| Median, months (95% CI)a | NE (NE, NE) | 11 (9, 13) |
| Intracranial overall response in patients with measurable CNS lesions at baseline | N=17 | N=13 |
| Intracranial response rate, n (%) | 14 (82%) | 3 (23%) |
| (95% CI)c | (57, 96) | (5, 54) |
| Complete response rate | 71% | 8% |
| Duration of response | ||
| Number of responders | 14 | 3 |
| Median, months (95% CI)a | NE (NE, NE) | 10 (9, 11) |
| Intracranial overall response in patients with any measurable or nonmeasurable CNS lesions at baseline | N=38 | N=40 |
| Intracranial response rate, n (%) | 25 (66%) | 8 (20%) |
| (95% CI)c | (49, 80) | (9, 36) |
| Complete response rate | 61% | 15% |
| Duration of response | ||
| Number of responders | 25 | 8 |
| Median, months (95% CI)a | NE (NE, NE) | 9 (6, 11) |
Abbreviations: BICR = blinded independent central review; CI = confidence interval; CNS = central nervous system; INV = investigator assessment; N/n = number of patients; NE = not estimable.
* p-value based on 1-sided stratified log-rank test.
a Based on the Brookmeyer and Crowley method.
b Hazard ratio based on Cox proportional hazards model; under proportional hazards, hazard ratio <1 indicates a reduction in hazard rate in favour of lorlatinib.
c Using exact method based on binomial distribution.
Figure 1. Kaplan-Meier plot of progression-free survival by blinded independent central review in CROWN study:
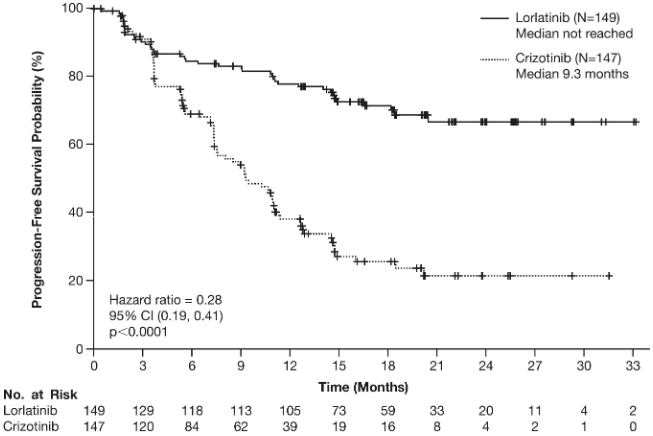
Abbreviations: CI=confidence interval; N/No.=number of patients.
The benefit from lorlatinib treatment was comparable across subgroups of baseline patient and disease characteristics, including patients with CNS metastases at baseline (n=38, HR=0.2, 95% CI: 0.10-0.43) and patients without CNS metastases at baseline (n=111, HR=0.32, 95% CI: 0.20-0.49).
ALK-positive advanced NSCLC previously treated with an ALK kinase inhibitor
The use of lorlatinib in the treatment of ALK-positive advanced NSCLC after treatment with at least one second-generation ALK TKI was investigated in Study A, a single-arm, multicentre Phase ½ study. A total of 139 patients with ALK-positive advanced NSCLC after treatment with at least one second-generation ALK TKI were enrolled in the Phase 2 portion of the study. Patients received lorlatinib orally at the recommended dose of 100 mg once daily, continuously.
The primary efficacy endpoint in the Phase 2 portion of the study was ORR, including intracranial (IC)-ORR, as per Independent Central Review (ICR) according to modified RECIST v1.1. Secondary endpoints included DOR, IC-DOR, time-to-tumour response (TTR) and PFS.
Patient demographics of the 139 ALK-positive advanced NSCLC patients after treatment with at least one second-generation ALK TKI, were 56% female, 48% White, 38% Asian, and the median age was 53 years (range: 29-83 years) with 16% of patients ≥65 years of age. The ECOG performance status at baseline was 0 or 1 in 96% patients. Brain metastases were present at baseline in 67% of patients. Of the 139 patients, 20% received 1 prior ALK TKI, excluding crizotinib, 47% received 2 prior ALK TKIs and 33% received 3 or more prior ALK TKIs.
The main efficacy results for Study A are included in Tables 4 and 5.
Table 4. Overall efficacy results in Study A by prior treatment:
| Efficacy parameter | One prior ALK TKIa with or without prior chemotherapy (N=28) | Two or more prior ALK TKIs with or without prior chemotherapy (N=111) |
|---|---|---|
| Objective response rateb (95% CI) Complete response, n Partial response, n | 42.9% (24.5, 62.8) 1 11 | 39.6% (30.5, 49.4) 2 42 |
| Duration of response Median, months (95% CI) | 5.6 (4.2, NR) | 9.9 (5.7, 24.4) |
| Progression-free survival Median, months (95% CI) | 5.5 (2.9, 8.2) | 6.9 (5.4, 9.5) |
Abbreviations: ALK = anaplastic lymphoma kinase; CI = confidence interval; ICR = Independent Central Review; N/n = number of patients; NR = not reached; TKI = tyrosine kinase inhibitor.
a Alectinib, brigatinib, or ceritinib.
b Per ICR.
Table 5. Intracranial* efficacy results in Study A by prior treatment:
| Efficacy parameter | One prior ALK TKIa with or without prior chemotherapy (N=9) | Two or more prior ALK TKIs with or without prior chemotherapy (N=48) |
|---|---|---|
| Objective response rateb (95% CI) Complete response, n Partial response, n | 66.7% (29.9, 92.5) 2 4 | 52.1% (37.2, 66.7) 10 15 |
| Duration of intra-cranial response Median, months (95% CI) | NR (4.1, NR) | 12.4 (6.0, NR) |
Abbreviations: ALK = anaplastic lymphoma kinase; CI = confidence interval; ICR = Independent Central Review; N/n = number of patients; NR = not reached; TKI = tyrosine kinase inhibitor.
* In patients with at least one measurable brain metastasis at baseline.
a Alectinib, brigatinib, or ceritinib.
b Per ICR.
In the overall efficacy population of 139 patients, 56 patients had a confirmed objective response by ICR with a median TTR of 1.4 months (range: 1.2 to 16.6 months). The ORR for Asians was 49.1% (95% CI: 35.1, 63.2) and 31.5% for non-Asians (95% CI: 21.1, 43.4). Among the 31 patients with a confirmed IC objective tumour response and at least one measurable brain metastasis at baseline by ICR, the median IC-TTR was 1.4 months (range: 1.2 to 16.2 months). The IC-ORR was 54.5% for Asians (95% CI: 32.2, 75.6) and 46.4% for non-Asians (95% CI: 27.5, 66.1).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with lorlatinib in all subsets of the paediatric population in lung carcinoma (small cell and non-small cell carcinoma) (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called ‘conditional approval’ scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
Pharmacokinetic properties
Absorption
Peak lorlatinib concentrations in plasma are rapidly reached with the median Tmax of 1.2 hours following a single 100 mg dose and 2.0 hours following multiple dosing of 100 mg once daily.
After oral administration of lorlatinib tablets, the mean absolute bioavailability is 80.8% (90% CI: 75.7, 86.2) compared to intravenous administration.
Administration of lorlatinib with a high fat, high calorie meal resulted in 5% higher exposure compared to fasted conditions. Lorlatinib may be administered with or without food.
At 100 mg once daily, the geometric mean (% coefficient of variation [CV]) peak plasma concentration was 577 (42) ng/mL and the AUC24 was 5,650 (39) ng h/mL in patients with cancer. The geometric mean (% CV) oral clearance was 17.7 (39) L/h.
Distribution
In vitro binding of lorlatinib to human plasma proteins is 66% with moderate binding to albumin or to α1-acid glycoprotein.
Biotransformation
In humans, lorlatinib undergoes oxidation and glucuronidation as the primary metabolic pathways. In vitro data indicate that lorlatinib is metabolised primarily by CYP3A4 and UGT1A4, with minor contribution from CYP2C8, CYP2C19, CYP3A5 and UGT1A3.
In plasma, a benzoic acid metabolite of lorlatinib resulting from the oxidative cleavage of the amide and aromatic ether bonds of lorlatinib was observed as a major metabolite, accounting for 21% of the circulating radioactivity. The oxidative cleavage metabolite is pharmacologically inactive.
Elimination
The plasma half-life of lorlatinib after a single 100 mg dose was 23.6 hours. The estimated lorlatinib effective plasma half-life at steady-state following completion of autoinduction was 14.83 hours. Following oral administration of a 100 mg radiolabelled dose of lorlatinib, a mean 47.7% of the radioactivity was recovered in urine and 40.9% of the radioactivity was recovered in faeces, with overall mean total recovery of 88.6%.
Unchanged lorlatinib was the major component of human plasma and faeces, accounting for 44% and 9.1% of total radioactivity, respectively. Less than 1% of unchanged lorlatinib was detected in urine.
Furthermore, lorlatinib is an inducer via human pregnane-X-receptor (PXR) and the human constitutive androstane receptor (CAR).
Linearity/non-linearity
At single dose, lorlatinib systemic exposure (AUCinf and Cmax) increased in a dose-related manner over the 10 to 200 mg dose range. Few data are available over the 10 to 200 mg dose range; however, no deviation from linearity was observed for AUCinf and Cmax after single dose.
After multiple once daily dose administration, lorlatinib Cmax increased dose-proportionally and AUCtau increased slightly less than proportionally over the dose range of 10 to 200 mg once daily.
Also, at steady-state lorlatinib plasma exposures are lower than those expected from single dose pharmacokinetics, indicative of a net time-dependent auto-induction effect.
Hepatic impairment
As lorlatinib is metabolised in the liver, hepatic impairment is likely to increase lorlatinib plasma concentrations. Clinical studies that were conducted excluded patients with AST or ALT >2.5 × ULN, or if due to underlying malignancy, >5.0 × ULN or with total bilirubin >1.5 × ULN. Population pharmacokinetic analyses have shown that lorlatinib exposure was not clinically meaningfully altered in patients with mild hepatic impairment (n=50). No dose adjustments are recommended for patients with mild hepatic impairment. No information is available for patients with moderate or severe hepatic impairment.
Renal impairment
Less than 1% of the administered dose is detected as unchanged lorlatinib in urine. Population pharmacokinetic analyses have shown that lorlatinib steady-state plasma exposure and Cmax values slightly increase with worsening baseline renal function. Based on a renal impairment study, no starting dose adjustments are recommended for patients with mild or moderate renal impairment [eGFR based on Modification of Diet in Renal Disease Study equation (MDRD)-derived eGFR (in mL/min/1.73 m²) × measured body surface area/1.73 ≥30 mL/min]. In this study, lorlatinib AUCinf increased by 41% in subjects with severe renal impairment (absolute eGFR <30 mL/min) compared to subjects with normal renal function (absolute eGFR ≥90 mL/min). A reduced dose of lorlatinib is recommended in patients with severe renal impairment, e.g., a once daily oral starting dose of 75 mg (see section 4.2). No information is available for patients on renal dialysis.
Age, gender, race, body weight and phenotype
Population pharmacokinetic analyses in patients with advanced NSCLC and healthy volunteers indicate that there are no clinically relevant effects of age, gender, race, body weight and phenotypes for CYP3A5 and CYP2C19.
Cardiac electrophysiology
In Study A, 2 patients (0.7%) had absolute Fridericia’s correction QTc (QTcF) values >500 msec and 5 patients (1.8%) had a change in QTcF from baseline >60 msec.
In addition, the effect of a single oral dose of lorlatinib (50 mg, 75 mg, and 100 mg) with and without 200 mg once daily itraconazole was evaluated in a 2-way crossover study in 16 healthy volunteers. No increases in the mean QTc were observed at the mean observed lorlatinib concentrations in this study.
In 295 patients who received lorlatinib at the recommended dose of 100 mg once daily and had a ECG measurement in Study A, lorlatinib was studied in a population of patients that excluded those with QTc interval >470 msec. In the study population, the maximum mean change from baseline for PR interval was 16.4 msec (2-sided 90% upper CI 19.4 msec) (see sections 4.2, 4.4 and 4.8). Of these, 7 patients had a baseline PR >200 msec. Among the 284 patients with PR interval <200 msec, 14% had PR interval prolongation ≥200 msec after starting lorlatinib. The prolongation of PR interval occurred in a concentration-dependent manner. Atrioventricular block occurred in 1.0% of patients.
For those patients who develop PR prolongation, dose modification may be required (see section 4.2).
Preclinical safety data
Repeat-dose toxicity
The main toxicities observed were inflammation across multiple tissues (skin and cervix of rats and lung, trachea, skin, lymph nodes and/or the oral cavity including mandibular bone of dogs; associated with increases in white blood cells, fibrinogen and/or globulin and decreases in albumin) and changes in the pancreas (with increases in amylase and lipase), hepatobiliary system (with increases in liver enzymes), male reproductive system, cardiovascular system, kidneys and gastrointestinal tract, peripheral nerves and the CNS (potential for cognitive functional impairment) at dose equivalent to human clinical exposure at the recommended posology. Changes in blood pressure and heart rate, and QRS complex and PR interval were also observed in animals after acute dosing (approximately 2.6 times the human clinical exposure at 100 mg after a single dose based on Cmax). All target organ findings with the exception of hepatic bile duct hyperplasia were partially to fully reversible.
Genotoxicity
Lorlatinib is not mutagenic but is aneugenic in vitro and in vivo with a no observed effect level for aneugenicity approximately 16.5 times human clinical exposure at 100 mg based on AUC.
Carcinogenicity
Carcinogenicity studies have not been conducted with lorlatinib. Reproductive toxicity Seminiferous tubular degeneration and/or atrophy in the testes, and epididymal changes (inflammation and/or vacuolation) were observed in the rat and dog. In the prostate, minimal to mild glandular atrophy was observed in dogs at dose equivalent to human clinical exposure at the recommended posology). The effects on male reproductive organs were partially to fully reversible.
In embryo-foetal toxicity studies, conducted in rats and rabbits, respectively, increased embryolethality and lower foetal body weights and malformations were observed. Foetal morphologic abnormalities included rotated limbs, supernumerary digits, gastroschisis, malformed kidneys, domed head, high arched palate and dilation of ventricles of the brain. The exposure at the lowest doses with embryo-foetal effects in animals was equivalent to the human clinical exposure at 100 mg, based on AUC.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.