LUPKYNIS Soft capsule Ref.[50195] Active ingredients: Voclosporin
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Otsuka Pharmaceutical Netherlands B.V., Herikerbergweg 292, 1101 CT Amsterdam, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, calcineurin inhibitors
ATC code: L04AD03
Mechanism of action
Voclosporin is a calcineurin-inhibitor immunosuppressant that inhibits calcineurin in a dose-dependent manner up to a maximum dose of 1.0 mg/kg. Activation of lymphocytes involves an increase in intracellular calcium concentrations. Calcineurin is a calcium/calmodulin-dependent phosphatase whose activity is required for the induction of T-cell lymphokine production and proliferation. The immunosuppressant activity results in inhibition of lymphocyte proliferation, T-cell cytokine production, and expression of T-cell activation surface antigens.
Pharmacodynamic effects
Cardiac electrophysiology
In a randomised, placebo- and active-controlled (moxifloxacin 400 mg), single dose study with parallel study design, dose-dependent QT prolonging effect was detected with voclosporin in the dose range of 0.5 mg/kg to 4.5 mg/kg (up to 9-fold coverage of the therapeutic exposure). Dose-dependent QT prolongation effect was observed with a time to maximum QTc increase occurring at 4 hours to 6 hours post-dose across different dose levels. The maximum mean placebo-adjusted changes of QTcF from baseline after voclosporin 0.5 mg/kg, 1.5 mg/kg, 3.0 mg/kg, and 4.5 mg/kg dose were 6.4 msec, 17.5 msec, 25.7 msec, and 34.6 msec, respectively.
In a separate, randomised, placebo-controlled, crossover study in 31 healthy subjects, an absence of large mean increases (i.e., >20 msec) was observed following 7 days of treatment with voclosporin at 0.3 mg/kg, 0.5 mg/kg and 1.5 mg/kg twice daily (approximately 6-fold coverage of the therapeutic exposure). The mechanism for the QT prolonging effect as observed in the single-dose and multipledose studies is unknown.
Based on data in LN patients receiving voclosporin 23.7 mg or 39.5 mg twice daily, a regression analysis of placebo corrected QTcF change from baseline showed a minimal negative slope (−0.065344 msec/ng/mL), not statistically different from a slope of 0 (p=0.1042).
Clinical efficacy and safety
The safety and efficacy of voclosporin were investigated in two placebo-controlled clinical trials (AURORA 1 and AURA-LV) in patients with LN of Class III or IV (alone or in combination with Class V) or pure Class V. All patients received background therapy of MMF (2 g/day) and corticosteroids (up to a total of 1 g of intravenous (IV) methylprednisolone over days 1 and 2 followed by a starting dose of oral corticosteroids of 25 mg/day (or 20 mg/day if body weight was <45 kg), tapered down to 2.5 mg/day by week 16.
Patients that completed the AURORA 1 study could continue in a 2-year continuation study (AURORA 2).
Phase 3 AURORA 1
The AURORA 1 study was a phase 3, prospective, randomised, double-blind, study comparing 23.7 mg (corresponding to a 0.37 mg/kg dose) twice daily of voclosporin (n=179) vs. placebo (n=178) over a 52-week treatment period. The demographic characteristics of patients in the study were well balanced across the two treatment arms. The mean age was 33 years (range 18 years to 72 years) and the majority of patients were female (87.7%), of which 81.8% were of childbearing potential.
Most patients were White (36.1%) or Asian (30.5%), and approximately one third of the study population was Hispanic or Latino. The mean weight was 66.5 kg (range 36 kg to 142 kg). The median time since systemic lupus erythematosus (SLE) diagnosis was 5.0 years and the median time since LN diagnosis was 2.0 years.
Before entering the AURORA 1 study, most patients (98%) had received treatment for LN in the past, with approximately 55% of patients taking MMF at screening. The proportion of LN treatment naïve patients was very low (2%).
More patients in the voclosporin arm than the placebo arm achieved the primary endpoint of renal response (table 4).
Table 4. AURORA 1 – Summary of key efficacy endpoints:
| Voclosporin (n=179) n (%) | Placebo (n=178) n (%) | Odds ratio vs. placebo (95 % CI) | p-value | |
|---|---|---|---|---|
| Renal response at week 52 | 73 (40.8) | 40 (22.5) | 2.65 (1.64, 4.27) | <0.001 |
| Renal response at week 24 | 58 (32.4) | 35 (19.7) | 2.23 (1.34, 3.72) | = 0.002 |
| Partial renal response* at week 24 | 126 (70.4) | 89 (50.0) | 2.43 (1.56, 3.79) | <0.001 |
| Partial renal response* at week 52 | 125 (69.8) | 92 (51.7) | 2.26 (1.45, 3.51) | <0.001 |
* Partial renal response defined as a 50% reduction in UPCR.
Notes: CI = Confidence interval; UPCR = Urine protein to creatinine ratio
The overall proportion of patients that achieved each of the components assessed for the primary endpoint at 52 weeks in the voclosporin vs placebo arm were:
- urine protein to creatinine ratio (UPCR) ≤0.5 mg/mg: 45.3% vs 23.0%
- with normal, stable renal function (defined as eGFR ≥60 mL/min/1.73 m² or no confirmed decrease from baseline in eGFR of >20%): 82.1% vs. 75.8%
- in the presence of sustained, low-dose steroids (not more than 10 mg for ≥3 consecutive days or for ≥7 days in total during weeks 44 to 52): 87.2% vs. 85.4%
- and received no rescue medication for LN: 91.1% vs. 86.5%
More patients in the voclosporin arm than the placebo arm achieved UPCR ≤0.5 mg/mg (64.8% vs. 43.8%) and the time to UPCR ≤0.5 mg/mg was significantly shorter for voclosporin treatment (median time: 169 days vs. 372 days for placebo treatment; hazard ratio (HR) 2.02; 95% CI: 1.51, 2.70; p<0.001).
Figure 1. Kaplan-Meier curve of time (days) to UPCR ≤0.5 mg/mg:
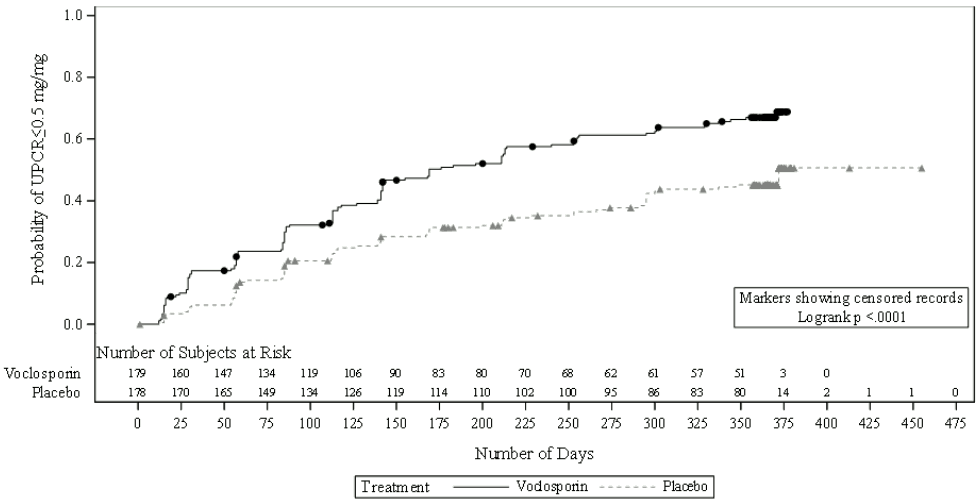
The time taken to reach a 50% reduction in UPCR was significantly shorter for the voclosporin arm than the placebo arm (HR 2.05; 95% CI: 1.62, 2.60; p<0.001). Median time to 50% reduction in UPCR was 29 days for voclosporin vs. 63 days for placebo (figure 2).
Figure 2. Kaplan-Meier curve of time (days) to 50% reduction in UPCR from baseline:
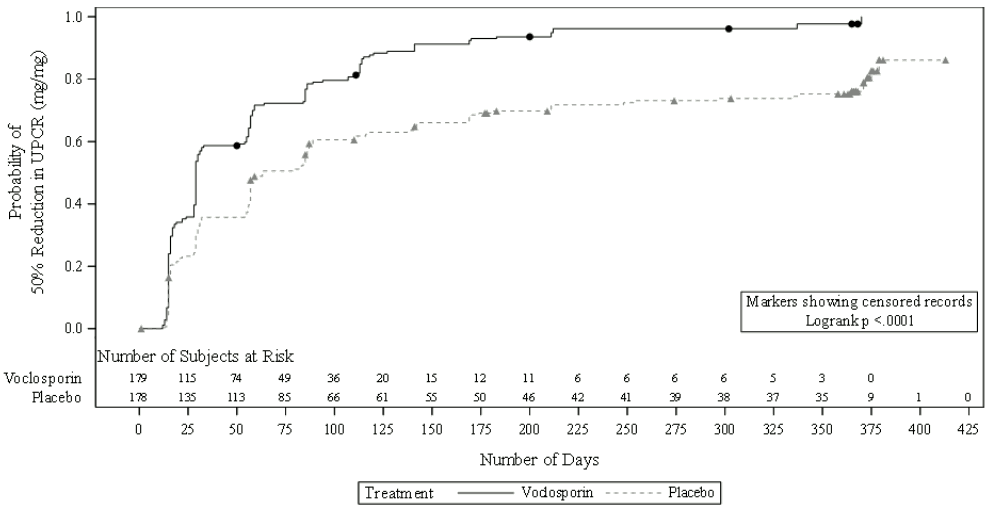
Over 80% of patients in the AURORA 1 study achieved a reduction in dose of oral corticosteroid to ≤2.5 mg/day at week 24 and this dose was maintained by over 75% of patients at week 52.
Phase 3 AURORA 2
The AURORA 2 study was a continuation study to evaluate the long-term safety and efficacy of voclosporin in patients that completed treatment in the AURORA 1 study. Patients stayed on the same treatment and dose of voclosporin (n=116) or placebo (n=100) as at the end of AURORA 1 and continued treatment for up to a further 2 years. Over 85% of patients completed the study (voclosporin: 87.1%, placebo 85.0%); 79.3% of voclosporin patients and 73% of placebo patients were still on study treatment at the end of study.
The proportion of patients in renal response at month 36 was 33% (59/179) in the voclosporin group and 22% (39/178) in the placebo group (ITT, AURORA 1) and 51% (59/116) in the voclosporin group and 39% (39/100) in the placebo group (ITT, AURORA 2).
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Lupkynis in one or more subsets of the paediatric population, in LN (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following oral administration (voclosporin 23.7 mg twice daily) the median time to reach maximum whole blood concentrations (Cmax) is 1.5 hours (range: 0.75 hour to 2 hours). With a twice daily dosing regimen, voclosporin steady state is achieved after 6 days and voclosporin accumulates approximately 2-fold relative to a single dose. At steady state, the whole blood mean Cmax and pre-dose trough values for voclosporin were 120 ng/mL (32% CV) and 15.0 ng/mL (49% CV), respectively. In vitro data investigating if voclosporin is a substrate of the efflux transporters P-gp or BCRP are inconclusive, but clinically relevant effects of P-gp/BCRP inhibitors are not expected.
Co-administration of voclosporin with food decreased both the rate and extent of absorption. Cmax and AUC of voclosporin were reduced by 53% and 25% when given with high-fat food and by 29% and 15% when given with low-fat food. These changes were not considered to be clinically relevant. Therefore, voclosporin can be taken with or without food.
Distribution
Voclosporin is 97% bound to plasma proteins. Voclosporin partitions extensively into red blood cells and distribution between whole blood and plasma is concentration- and temperature-dependent. A population pharmacokinetic analysis in patients resulted in an apparent volume of distribution (Vss/F) of 2,154 L.
Biotransformation
Voclosporin is extensively metabolised, predominantly by CYP3A4 to form oxidative metabolites. Voclosporin is the major circulating component following a single dose of [14C]-voclosporin. One major metabolite was observed in human whole blood and represented 16.7% of total exposure. The major metabolite is not expected to contribute to the pharmacological activity of voclosporin since it was reported as about 8-fold less potent in a lymphocyte proliferation assay and has lower exposure than voclosporin.
Elimination
The mean apparent clearance at steady state (CLss/F) after voclosporin 23.7 mg twice daily is 63.6 L/h (37.5% CV). The mean terminal half-life (t½) at steady state is approximately 30 hours (range: 24.9 hours to 36.5 hours).
Following single oral administration of 70 mg [14C]-voclosporin, 94.8% of the radioactivity was recovered by 168 hours post dose: 92.7% was recovered in faeces (including 5% as unchanged voclosporin), and 2.1% was recovered in urine (including 0.25% as unchanged voclosporin).
In-vitro data indicate that voclosporin is an inhibitor of OATP1B1 and OATP1B3.
Linearity/non-linearity
In healthy volunteers, a non-linearity between dose and exposure was observed at the lower end of the dose range studied (0.25 mg/kg to 1.5 mg/kg twice daily), which had a relatively minor effect on the pharmacokinetics. The dose-proportionality factor was always less than 1.5. This non-linearity has not been detected over the dose range studied in LN patients.
Pharmacokinetics in special populations
Renal impairment
In clinical studies, kidney function was monitored by eGFR and doses were adjusted based on a predefined dose adjustment protocol. Enrolled LN patients had a baseline eGFR > 45 mL/min/1.73 m².
Dosing adjustments have to follow the recommendations outlined in table 1. A dedicated renal impairment study revealed that after single and multiple doses of voclosporin, Cmax and AUC were similar in volunteers with mild (creatinine clearance (CLCr) 60 mL/min to 89 mL/min as estimated by Cockcroft Gault) and moderate (CLCr 30 mL/min to 59 mL/min) renal impairment compared to volunteers with normal renal function (CLCr ≥90 mL/min). After a single dose of voclosporin in volunteers with severe renal impairment (CLCr <30 mL/min), Cmax and AUC increased 1.5-fold and 1.7-fold, respectively. The effect of end-stage renal disease (ESRD) with or without haemodialysis on the pharmacokinetics of voclosporin is unknown (see section 4.2).
Hepatic impairment
A dedicated hepatic impairment study compared systemic exposure of voclosporin in patients with mild or moderate hepatic impairment (Child-Pugh A and B, respectively) vs. healthy controls with normal hepatic function. In patients with mild and moderate hepatic impairment, voclosporin Cmax and AUC0-48 increased by 1.5-fold and approximately 2-fold, respectively (see section 4.2). Voclosporin has not been evaluated in patients with severe hepatic impairment (Child-Pugh C) and its use in these patients is not recommended (see section 4.4).
Age, sex, race and body weight
A population pharmacokinetic analysis assessing the effects of age, sex, race and body weight did not suggest any clinically significant impact of these covariates on voclosporin exposures.
5.3. Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:
Repeated-dose animal studies have shown neuro-histological findings of gliosis and perivascular infiltrates in the brain and spinal cord in rats, but not in dogs or monkeys. These findings were not observed at doses approximately 0.3-times the maximum recommended human dose (MRHD) of 23.7 mg voclosporin twice a day on medicinal product exposure (AUC) basis.
In a 39-week oral toxicology study with cynomolgus monkeys, malignant lymphomas occurred at a dose of 150 mg/kg/day (approximately 4-times and 7-times above the MRHD based on medicinal product exposure (AUC), for male and female animals, respectively). At this dose, monkeys experienced high levels of immunosuppression as indicated by maximum calcineurin inhibition levels (Emax) of greater than 80 %. The no-observed-adverse-effect level (NOAEL) for this finding was 75 mg/kg/day (approximately 4-times the MRHD, on medicinal product exposure (AUC) basis, for male and female animals).
No mutagenic or genotoxic effects of voclosporin were observed in conventional genotoxicity studies.
In a 2-year mouse carcinogenicity study with oral voclosporin, an increased incidence of malignant lymphoma was observed at the highest dose tested (30 mg/kg/day; approximately 7.5-times the MRHD on a medicinal product exposure (AUC) basis). This result is considered secondary to voclosporin-related immune suppression. The NOAEL was 10 mg/kg/day (approximately 1-times the MRHD on medicinal product exposure (AUC) basis).
In a rat fertility study with a 50:50 mixture of voclosporin and its cis-isomer, decreases in male reproductive organ weights, including the cauda epididymis, epididymis, seminal vesicles, prostate, and testes were noted at a dose of 25 mg/kg/day. The NOAEL for these findings was 10 mg/kg/day (approximately 5-times the MRHD on medicinal product exposure (AUC) basis). Mating and fertility parameters, sperm motility, count and density, number of estrous stages per 14 days, and caesarean sectioning parameters were not affected. Decreases in prostate and testes weights were also observed in the 13-week and 26-week repeat-dose toxicity studies with oral 50:50 mixture of voclosporin and its cis-isomer at doses of 25 mg/kg/day and 10 mg/kg/day, or 18-times and 7-times the MRHD, on medicinal product exposure (AUC) basis. The NOAEL for these effects in the 26-week repeat-dose study was 2.5 mg/kg/day (approximately 1-times the MRHD on medicinal product exposure (AUC) basis).
Embryo-foetal development studies were conducted with the 50:50 mixture of voclosporin and its cisisomer in both rats and rabbits and with voclosporin in rabbits. Embryo-foetal toxicity was only observed at doses that were associated with maternal toxicity (at doses approximately 15-times and 1-times the MRHD, based upon medicinal product exposure (AUC), for rats and rabbits, respectively). The maternal effects included changes in body weight and/or swollen mammary glands while the foetal effects consisted of a slight reduction in body weight and related skeletal developmental variations. No malformative effects were noted in the studies. The NOAELs were 10 mg/kg/day in rats and 1 mg/kg/day in rabbits (approximately 7-times and 0.01-times the MRHD, based on medicinal product exposure (AUC), for rats and rabbits, respectively).
In a pre- and post-natal developmental study in rats, maternal toxicity at a dose of 25 mg/kg/day 50:50 mixture of voclosporin and its cis-isomer (approximately 17 times the MRHD on medicinal product exposure (AUC) basis) delayed parturition (dystocia) that resulted in reductions in the mean number of total pups delivered and surviving pups per litter. This dose was associated with maternal toxicity based on decreased body weight gain. No adverse effects on dams or their pups were observed at doses approximately 3-times the MRHD and lower (based on medicinal product exposure (AUC) with a maternal oral NOAEL dose of 10 mg/kg/day). There were no effects on behavioural and physical development, or the reproductive performance of male or female pups. The no effect dose for delivery and pup survival was 10 mg/kg/day.
Medicinal product-derived radioactivity was rapidly distributed to milk following the oral administration of [14C]-voclosporin to lactating rats. When a medicinal product is present in animal milk, it is likely that it will also be present in human milk.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.