NERLYNX Film-coated tablet Ref.[110548] Active ingredients: Neratinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: PIERRE FABRE MEDICAMENT, Les Cauquillous, 81500 Lavaur, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors
ATC code: L01EH02
Mechanism of action
Neratinib is an irreversible pan–erythroblastic leukaemia viral oncogene homolog (ERBB) tyrosine kinase inhibitor (TKI) that blocks mitogenic growth factor signal transduction through covalent, high affinity binding to the ATP binding site of 3 epidermal growth factor receptors (EGFRs): EGFR (encoded by ERBB1), HER2 (encoded by ERBB2), and HER4 (encoded by ERBB4) or their active heterodimers with HER3 (encoded by ERBB3). This results in sustained inhibition of these growth promoting pathways with HER2-amplified or over-expressed, or HER2-mutant breast cancers. Neratinib binds to the HER2 receptor, reduces EGFR and HER2 autophosphorylation, downstream MAPK and AKT signaling pathways, and potently inhibits tumour cell proliferation in vitro. Neratinib inhibited EGFR and/or HER2-expressing carcinoma cell lines with a cellular IC50 <100 nM.
Clinical efficacy and safety
In the multicentre, randomised, double-blind, placebo-controlled, pivotal phase III study, ExteNET (3004), 2 840 women with early-stage HER2-positive breast cancer (as confirmed locally by assay) who had completed adjuvant treatment with trastuzumab were randomised 1:1 to receive either Nerlynx or placebo daily for one year. The median age in the intention-to-treat (ITT) population was 52 years (59.9% was ≥50 years old, 12.3% was ≥65 years old); 81.0% were Caucasian, 2.6% black or African American, 13.6% Asian and 2.9% other. At baseline, 57.7% had hormone receptor positive disease (defined as ER-positive and/or PgR-positive), 27.2% were node negative, 41.5% had one to three positive nodes and 29.4% had four or more positive nodes. Approximately 10% of patients had Stage I tumours, approximately 40% had Stage II tumours and approximately 30% had Stage III tumours. Median time from the last adjuvant trastuzumab treatment to randomization was 4.5 months.
The primary endpoint of the study was invasive disease-free survival (iDFS). Secondary endpoints of the study included disease-free survival (DFS) including ductal carcinoma in situ (DFS-DCIS), time to distant recurrence (TTDR), distant disease-free survival (DDFS), cumulative incidence of central nervous system recurrence and overall survival (OS).
The primary analysis of the study after 2 years post-randomisation demonstrated that Nerlynx significantly reduced the risk of invasive disease recurrence or death by 33% (HR=0.67 with 95% CI (0.49, 0.91), two-sided p=0.011) in the ITT population.
Table 6. Primary 2-year efficacy results – ITT and hormone receptor positive populations who were less than one year from completion of trastuzumab therapy:
| Variable | Estimated 2 year event free rates1 (%) | Hazard ratio (95% CI)2 | P-value3 | |
|---|---|---|---|---|
| ITT population | ||||
| Nerlynx (N=1420) | Placebo (N=1420) | |||
| Invasive disease-free survival | 94.2 | 91.9 | 0.67 (0.49, 0.91) | 0.011 |
| Disease-free survival including ductal carcinoma in situ | 94.2 | 91.3 | 0.62 (0.46, 0.84) | 0.002 |
| Distant disease-free survival | 95.3 | 94.0 | 0.75 (0.53, 1.06) | 0.110 |
| Time to distant recurrence | 95.5 | 94.2 | 0.74 (0.52, 1.06) | 0.102 |
| CNS recurrence | 0.92 | 1.16 | – | 0.586 |
| Hormone receptor positive population who were less than one year from completion of trastuzumab | ||||
| Nerlynx (N=671) | Placebo (N=668) | Hazard ratio (95% CI)4 | P-value5 | |
| Invasive disease-free survival | 95.3 | 90.9 | 0.50 (0.31, 0.78) | 0.003 |
| Disease-free survival including ductal carcinoma in situ | 95.3 | 90.1 | 0.45 (0.28, 0.71) | <0.001 |
| Distant disease-free survival | 96.1 | 93.0 | 0.53 (0.31, 0.88) | 0.015 |
| Time to distant recurrence | 96.3 | 93.3 | 0.53 (0.30, 0.89) | 0.018 |
| CNS recurrence | 0.34 | 1.01 | – | 0.189 |
CNS = central nervous system.
1 Event-free rates for all endpoints, except for CNS recurrence for which cumulative incidence is reported.
2 Stratified Cox proportional hazards model
3 Stratified 2-sided log-rank test for all endpoints, except for CNS recurrence for which Gray's method was used.
4 Unstratified Cox proportional hazards model 5 Unstratified 2-sided log-rank test for all endpoints, except for CNS recurrence for which Gray's method was used.
Figure 1. Kaplan-Meier plot of invasive disease-free survival – hormone receptor positive population who were less than one year from completion of trastuzumab therapy:
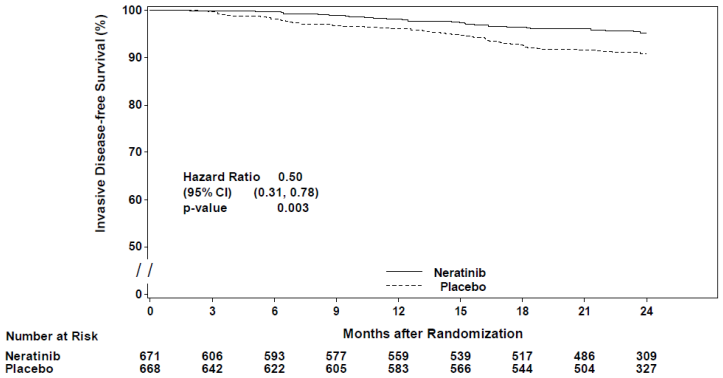
For hormone receptor positive patients who were less than one year from completion of trastuzumab therapy, the relative treatment benefit of Nerlynx within pre-specified patient subgroups is presented in Figure 2.
Figure 2. Hormone receptor positive patients who were less than one year from completion of trastuzumab therapy, invasive disease-free survival by patient subgroup:
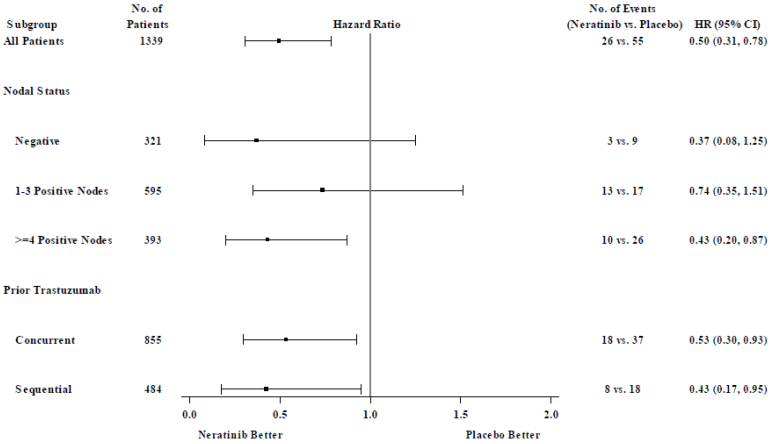
Note: Patients (n=30) who had an unknown nodal status are not shown because the HR could not be estimated
In patients that were hormone receptor negative, regardless of time from trastuzumab therapy, the hazard ratio for iDFS at 2 years was 0.94, with 95% CI (0.61, 1.46). In this population, efficacy has not been demonstrated.
Approximately 75% of patients were re-consented for extended follow-up beyond 24 months. Observations with missing data were censored at the last date of assessment. While the treatment benefit of Nerlynx over placebo was maintained at five years, the effect size cannot be reliably estimated.
The median OS follow-up time for the ITT population was 8.06 years, 8.03 years in the neratinib arm and 8.10 years in the placebo arm, with a total of 1542 (54.3%) patients followed up for survival for 8 or more years, 746 (52.5%) in the neratinib arm and 796 (56.1%) in the placebo arm. The number of deaths was 264 (9.3%), with 127 (8.9%) in the patients treated with neratinib and 137 (9.6%) in the patients treated with placebo.
There was no statistically significant difference in overall survival between the Nerlynx and the placebo arm [HR 0.96 (95% CI: 0.75, 1.22)] in the ITT population at a median follow-up of 8.06 years.
In the hormone receptor positive population who were less than one year from completion of trastuzumab therapy, the median follow-up was 8.0 years in the neratinib arm and 8.1 years in the placebo arm, with a total of 1 339 (47.1%) patients followed up for survival for 8 or more years, 671 (23.6%) in the neratinib arm and 668 (23.5%) in the placebo arm. In this subpopulation the number of deaths was 55 (8.2%) in the patients treated with neratinib and 68 (10.2%) in the patients treated with placebo [HR 0.83 (95% CI, 0.58, 1.18)].
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies in all subsets of the paediatric population in the treatment of breast carcinoma.
5.2. Pharmacokinetic properties
The mass balance after administration of a single oral dose of 200 mg of neratinib was studied in six healthy subjects.
Absorption
Following oral administration of 240 mg neratinib, absorption was slow and peak plasma concentrations of neratinib occurred around 7 hours after administration. A single dose of 240 mg neratinib taken with food increased Cmax and AUC by approximately 17% and 13%, respectively, compared with administration in the fasting state. A single oral dose of 240 mg neratinib taken with a meal high in fat increased both Cmax and AUC by approximately 100%. In a mass balance study, the total recovery (urinary and fecal excretion) of intact neratinib and metabolites demonstrates that the fraction absorbed for neratinib is at least 10% and likely more than 20%. Moreover, model-based predictions suggested an overall absorbed fraction from the gut (fa) of 26%.
In vitro neratinib solubility is pH-dependent. Treatments that increase gastrointestinal pH may lower the absorption of neratinib, thus decreasing systemic exposure.
Distribution
Binding of neratinib to human plasma proteins, including covalent binding to human serum albumin (HSA), was greater than 98% and independent of the tested neratinib concentration. Neratinib bound predominantly to HSA and human alpha-1 acid glycoprotein (AAG). Binding of M6 main metabolite (M6) to human plasma proteins was greater than 99% and independent of the tested M6 concentrations.
In vitro studies demonstrated that neratinib is a substrate for P-glycoprotein (P-gp) (see sections 4.2, 4.3, 4.4 and 4.5) and BCRP. In vitro studies demonstrated that neratinib and its main metabolite M6 are not substrates of hepatic uptake transporters OATP1B1*1a and OATP1B3 at relevant clinical concentration.
Biotransformation
Neratinib is metabolised primarily in liver microsomes by CYP3A4 and to a lesser extent by flavincontaining monooxygenase (FMO).
Preliminary metabolite profiling in human plasma indicates that after oral administration, neratinib undergoes oxidative metabolism through CYP3A4. Circulating metabolites include neratinib pyridine N-oxide (M3), N-desmethyl neratinib (M6), neratinib dimethylamine N-oxide (M7) and traces of hydroxyl neratinib N-oxide and neratinib bis-N-oxide (M11). Neratinib represents the most prominent component in plasma and amongst circulating metabolites (M2, M3, M6, M7 and M11) none is above 8% of neratinib plus metabolite total exposure after oral administration of neratinib. The neratinib metabolites M3, M6, M7 and M11 were shown to have similar potencies to neratinib in either in vitro enzyme (binding assays) or cell based assays against cells expressing ERBB1, ERBB2 (HER2) and ERBB4.
Based on steady state exposures, neratinib provides the majority of pharmacological activity (73%), with 20% provided by exposure to M6, 6% provided by M3, and negligible contribution (<1%) from M7 and M11 AUC.
Elimination
Following single doses of neratinib, the mean apparent plasma half-life of neratinib was 17 hours in patients.
Excretion of neratinib is primarily via the faeces
Following the administration of a single radiolabelled dose of 240 mg neratinib oral solution, 95.5% and 0.96% of the administered dose was recovered in the faeces and urine, respectively.
The excretion was rapid and complete, with most of the dose recovered in faeces within 48 hours and 96.5% of total radioactivity recovered in excreta after 8 days. Unchanged neratinib was the most abundant species in excreta accounting for 62.1% of total dose recovered in excreta. The most abundant metabolites in faeces were M6 (19.7% of administered dose), followed by M2, M3 and M7, all below 10% of administered dose.
Medicinal product interactions
Effect of CYP3A4/P-gp inducer on neratinib
Following concomitant administration of 240 mg neratinib with repeated doses of 600 mg rifampicin, a strong CYP3A4/P-gp inducer, neratinib exposures were significantly decreased by 76% and 87% for Cmax and AUC, respectively, compared with neratinib administration alone (see sections 4.3 and 4.5).
Effect of CYP3A4/P-gp inhibitor on neratinib
Co-administration of a single oral dose of 240 mg of neratinib in the presence of ketoconazole (400 mg once daily for 5 days), a strong CYP3A4/P-gp inhibitor, increased neratinib systemic exposure by 3.2- and 4.8-fold for Cmax and AUC, respectively, compared with neratinib administered alone.
Model-based predictions suggested that co-administration of a single oral dose of 240 mg of neratinib in the presence of fluconazole (200 mg once daily for 8 days), a moderate CYP3A4 inhibitor, increased neratinib systemic exposure by 1.3- and 1.7-fold for Cmax and AUC, compared with neratinib administered alone.
Model-based predictions suggested that co-administration of a single oral dose of 240 mg of neratinib in the presence of verapamil (120 mg twice daily for 8 days), a moderate CYP3A4/strong P-gp inhibitor, increased neratinib systemic exposure by 3.0- and 4.0-fold for Cmax and AUC, compared with neratinib administered alone (see sections 4.2, 4.4 and 4.5).
Effect of gastric pH modifiers on neratinib
Co-administration of lansoprazole or ranitidine (1x300 mg) with a 240 mg single dose of neratinib in healthy volunteers resulted in a decreased neratinib exposure by around 70% or 50%, respectively. The magnitude of ranitidine interaction on neratinib AUC was reduced by around 25%, by staggering the administration of ranitidine (2x150 mg) 2 hours after neratinib administration (see sections 4.2, 4.4 and 4.5).
Effect of other treatment on neratinib
There were no apparent clinically relevant drug-drug interactions observed for neratinib when administered concomitantly with capecitabine, paclitaxel, trastuzumab, vinorelbine, or antidiarrhoeals (loperamide) (see section 4.5). Effect of neratinib on CYP substrates Neratinib and metabolite M6 were not potent direct inhibitors of CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, or 3A4 and no time-dependent inhibition is expected. Neratinib did not induce CYP1A2, 2B6, 2C9, or 3A4.
Effect of neratinib on transporters
There was no clinically relevant inhibition of human BSEP efflux transporter activity in vitro, with a reported IC50 value of >10 µM. Neratinib at 10 µM appeared to inhibit the BCRP efflux transporter which could be clinically relevant at intestinal level (see section 4.5).
In in vitro studies, neratinib was an inhibitor of P-glycoprotein (P-gp) efflux transporters, which was further confirmed in a clinical study. Multiple oral doses of neratinib 240 mg increased digoxin exposures (54 and 32% increase in Cmax and AUC, respectively) with no impact on its renal clearance level (see sections 4.4 and 4.5).
Neratinib produced no inhibitory activity towards the uptake transporters, OATP1B1*1a, OATP1B3, OAT1, OAT3 and OCT2, with reported IC50 values were >10µM. Neratinib produced inhibitory activity in OCT1 uptake transporter, with an IC50 of 2.9 µM.
Special populations
Renal impairment
Pharmacokinetic studies in patients with renal impairment or undergoing dialysis have not been carried out. Population pharmacokinetic modelling revealed that creatinine clearance did not explain the variability between patients, hence, no dose modifications are recommended for patients with mild to moderate renal impairment (see sections 4.2 and 4.4).
Hepatic impairment
Neratinib is extensively metabolised in the liver. In subjects with severe pre-existing hepatic impairment (Child-Pugh Class C) without cancer, the clearance of neratinib was decreased by 36% and exposure to neratinib increased by about 3-fold as compared to healthy volunteers (see sections 4.2 and 4.3).
5.3. Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:
Carcinogenesis, mutagenesis
Nerlynx was neither clastogenic nor mutagenic in the standard battery of genotoxicity studies.
Neratinib metabolites M3, M6, M7 and M11 are negative in the standard battery of in vitro genotoxicity studies.
A 6-month carcinogenicity study in Tg.rasH2 transgenic mice and the rat 2-year data showed no signs of carcinogenic potential.
Reproductive toxicity
In rabbits, there were no effects on mating or the ability of animals to become pregnant, but embryofoetal lethality and foetal morphologic anomalies (e.g. domed head, dilation of brain ventricles and misshapen anterior fontanelles and enlarged anterior and/or posterior fontanelles) were observed at doses that may be considered to be clinically relevant.
Environmental risk assessment (ERA)
Environmental risk assessment studies have shown that neratinib has an evident potential to be persistent, bioaccumulative, and toxic to the environment (see section 6.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.