NEVIRAPINE TEVA Tablet Ref.[8787] Active ingredients: Nevirapine
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Teva B.V., Swensweg 5, 2031GA Haarlem, The Netherlands
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Readministration to patients who have required permanent discontinuation for severe rash, rash accompanied by constitutional symptoms, hypersensitivity reactions, or clinical hepatitis due to nevirapine.
Patients with severe hepatic impairment (Child-Pugh C) or pre-treatment ASAT or ALAT >5 ULN until baseline ASAT/ALAT are stabilised <5 ULN.
Readministration to patients who previously had ASAT or ALAT >5 ULN during nevirapine therapy and had recurrence of liver function abnormalities upon readministration of nevirapine (see section 4.4).
Coadministration with herbal preparations containing St John’s wort (Hypericum perforatum) due to the risk of decreased plasma concentrations and reduced clinical effects of nevirapine (see section 4.5).
Special warnings and precautions for use
Nevirapine Teva should only be used with at least two other antiretroviral agents (see section 5.1).
Nevirapine Teva should not be used as the sole active antiretroviral, as monotherapy with any antiretroviral has shown to result in viral resistance.
The first 18 weeks of therapy with nevirapine are a critical period which requires close monitoring of patients to disclose the potential appearance of severe and life-threatening skin reactions (including cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN)) and serious hepatitis/hepatic failure. The greatest risk of hepatic and skin reactions occurs in the first 6 weeks of therapy. However, the risk of any hepatic event continues past this period and monitoring should continue at frequent intervals. Female gender and higher CD4 counts (>250/mm³ in adult females and >400/mm³ in adult males) at the initiation of nevirapine therapy are associated with a greater risk of hepatic adverse reactions if the patient has detectable plasma HIV-1 RNA – i.e. a concentration ≥50 copies/ml – at the initiation of nevirapine. As serious and life threatening hepatotoxicity has been observed in controlled and uncontrolled studies predominantly in patients with a plasma HIV-1 viral load of 50 copies/ml or higher, nevirapine should not be initiated in adult females with CD4 cell counts greater than 250 cells/mm³ or in adult males with CD4 cell counts greater than 400 cells/mm³, who have a detectable plasma HIV-1 RNA unless the benefit outweighs the risk. In some cases, hepatic injury has progressed despite discontinuation of treatment. Patients developing signs or symptoms of hepatitis, severe skin reaction or hypersensitivity reactions must discontinue nevirapine and seek medical evaluation immediately. Nevirapine must not be restarted following severe hepatic, skin or hypersensitivity reactions (see section 4.3).
The dose must be strictly adhered to, especially the 14-days lead-in period (see section 4.2).
Cutaneous reactions
Severe and life-threatening skin reactions, including fatal cases, have occurred in patients treated with nevirapine mainly during the first 6 weeks of therapy. These have included cases of Stevens-Johnson syndrome, toxic epidermal necrolysis and hypersensitivity reactions characterised by rash, constitutional findings and visceral involvement. Patients should be intensively monitored during the first 18 weeks of treatment. Patients should be closely monitored if an isolated rash occurs. Nevirapine must be permanently discontinued in any patient experiencing severe rash or a rash accompanied by constitutional symptoms (such as fever, blistering, oral lesions, conjunctivitis, facial oedema, muscle or joint aches, or general malaise), including Stevens-Johnson syndrome, or toxic epidermal necrolysis. Nevirapine must be permanently discontinued in any patient experiencing hypersensitivity reaction (characterised by rash with constitutional symptoms, plus visceral involvement, such as hepatitis, eosinophilia, granulocytopenia, and renal dysfunction), see section 4.4.
Nevirapine Teva administration above the recommended dose might increase the frequency and seriousness of skin reactions, such as Stevens-Johnson syndrome and toxic epidermal necrolysis.
Rhabdomyolysis has been observed in patients experiencing skin and/or liver reactions associated with nevirapine use.
Concomitant prednisone use (40 mg/day for the first 14 days of nevirapine administration) has been shown not to decrease the incidence of nevirapine-associated rash, and may be associated with an increase in incidence and severity of rash during the first 6 weeks of nevirapine therapy.
Some risk factors for developing serious cutaneous reactions have been identified; they include failure to follow the initial dosing of 200 mg daily during the lead-in period and a long delay between the initial symptoms and medical consultation. Women appear to be at higher risk than men of developing rash, whether receiving nevirapine or non-nevirapine containing therapy.
Patients should be instructed that a major toxicity of nevirapine is rash. They should be advised to promptly notify their physician of any rash and avoid delay between the initial symptoms and medical consultation. The majority of rashes associated with nevirapine occur within the first 6 weeks of initiation of therapy. Therefore, patients should be monitored carefully for the appearance of rash during this period. Patients should be instructed that dose escalation is not to occur if any rash occurs during the two-week lead-in dosing period, until the rash resolves. The 200 mg once daily dosing regimen should not be continued beyond 28 days at which point in time an alternative treatment should be sought due to the possible risk of underexposure and resistance.
Any patient experiencing severe rash or a rash accompanied by constitutional symptoms such as fever, blistering, oral lesions, conjunctivitis, facial oedema, muscle or joint aches, or general malaise should discontinue the medicinal product and immediately seek medical evaluation. In these patients nevirapine must not be restarted.
If patients present with a suspected nevirapine-associated rash, liver function tests should be performed. Patients with moderate to severe elevations (ASAT or ALAT >5 ULN) should be permanently discontinued from nevirapine.
If a hypersensitivity reaction occurs, characterised by rash with constitutional symptoms such as fever, arthralgia, myalgia and lymphadenopathy, plus visceral involvement, such as hepatitis, eosinophilia, granulocytopenia, and renal dysfunction, nevirapine must be permanently stopped and not be reintroduced (see section 4.3).
Hepatic reactions
Severe and life-threatening hepatotoxicity, including fatal fulminant hepatitis, has occurred in patients treated with nevirapine. The first 18 weeks of treatment is a critical period which requires close monitoring. The risk of hepatic reactions is greatest in the first 6 weeks of therapy. However the risk continues past this period and monitoring should continue at frequent intervals throughout treatment.
Rhabdomyolysis has been observed in patients experiencing skin and/or liver reactions associated with nevirapine use.
Increased ASAT or ALAT levels ≥ 2.5 ULN and/or co-infection with hepatitis B and/or C at the start of antiretroviral therapy is associated with greater risk of hepatic adverse reactions during antiretroviral therapy in general, including nevirapine containing regimens.
Female gender and higher CD4 counts at the initiation of nevirapine therapy in treatment-naïve patients is associated with increased risk of hepatic adverse reactions. Women have a three fold higher risk than men for symptomatic, often rash-associated, hepatic events (5.8% versus 2.2%), and treatment-naïve patients of either gender with detectable HIV-1 RNA in plasma with higher CD4 counts at initiation of nevirapine therapy are at higher risk for symptomatic hepatic events with nevirapine. In a retrospective review of predominantly patients with a plasma HIV-1 viral load of 50 copies/ml or higher, women with CD4 counts >250 cells/mm³ had a 12 fold higher risk of symptomatic hepatic adverse reactions compared to women with CD4 counts <250 cells/mm³ (11.0% versus 0.9%). An increased risk was observed in men with detectable HIV-1 RNA in plasma and CD4 counts >400 cells/mm³ (6.3% versus 1.2% for men with CD4 counts <400 cells/mm³). This increased risk for toxicity based on CD4 count thresholds has not been detected in patients with undetectable (i.e. <50 copies/ml) plasma viral load.
Patients should be informed that hepatic reactions are a major toxicity of nevirapine requiring close monitoring during the first 18 weeks. They should be informed that occurrence of symptoms suggestive of hepatitis should lead them to discontinue nevirapine and immediately seek medical evaluation, which should include liver function tests.
Liver monitoring
Clinical chemistry tests, which include liver function tests, should be performed prior to initiating nevirapine therapy and at appropriate intervals during therapy.
Abnormal liver function tests have been reported with nevirapine, some in the first few weeks of therapy.
Asymptomatic elevations of liver enzymes are frequently described and are not necessarily a contraindication to use nevirapine. Asymptomatic GGT elevations are not a contraindication to continue therapy.
Monitoring of hepatic tests should be done every two weeks during the first 2 months of treatment, at the 3rd month and then regularly thereafter. Liver test monitoring should be performed if the patient experiences signs or symptoms suggestive of hepatitis and/or hypersensitivity.
If ASAT or ALAT ≥2.5 ULN before or during treatment, then liver tests should be monitored more frequently during regular clinic visits. Nevirapine must not be administered to patients with pre-treatment ASAT or ALAT >5 ULN until baseline ASAT/ALAT are stabilised <5 ULN (see section 4.3). nevirapine therapy and at appropriate intervals during therapy.
Physicians and patients should be vigilant for prodromal signs or findings of hepatitis, such as anorexia, nausea, jaundice, bilirubinuria, acholic stools, hepatomegaly or liver tenderness. Patients should be instructed to seek medical attention promptly if these occur.
If ASAT or ALAT increase to >5 ULN during treatment, nevirapine should be immediately stopped. If ASAT and ALAT return to baseline values and if the patient had no clinical signs or symptoms of hepatitis, rash, constitutional symptoms or other findings suggestive of organ dysfunction, it may be possible to reintroduce nevirapine, on a case by case basis, at the starting dose regimen of 200 mg/day for 14 days followed by 400 mg/day. In these cases, more frequent liver monitoring is required. If liver function abnormalities recur, nevirapine should be permanently discontinued.
If clinical hepatitis occurs, characterised by anorexia, nausea, vomiting, icterus AND laboratory findings (such as moderate or severe liver function test abnormalities (excluding GGT)), nevirapine must be permanently stopped. Nevirapine must not be readministered to patients who have required permanent discontinuation for clinical hepatitis due to nevirapine.
Liver disease
The safety and efficacy of nevirapine has not been established in patients with significant underlying liver disorders. Nevirapine is contraindicated in patients with severe hepatic impairment (Child-Pugh C, see section 4.3). Pharmacokinetic results suggest caution should be exercised when nevirapine is administered to patients with moderate hepatic dysfunction (Child-Pugh B). Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reactions. In the case of concomitant antiviral therapy for hepatitis B or C, please refer also to the relevant product information for these medicinal products.
Patients with pre-existing liver dysfunction including chronic active hepatitis have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
Other warnings
Post-Exposure-Prophylaxis
Serious hepatotoxicity, including liver failure requiring transplantation, has been reported in HIV-uninfected individuals receiving multiple doses of nevirapine in the setting of post-exposure-prophylaxis (PEP), an unapproved use. The use of nevirapine has not been evaluated within a specific study on PEP, especially in term of treatment duration and therefore, is strongly discouraged.
Combination therapy with nevirapine is not a curative treatment of patients infected with HIV-1; patients may continue to experience illnesses associated with advanced HIV-1 infection, including opportunistic infections.
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
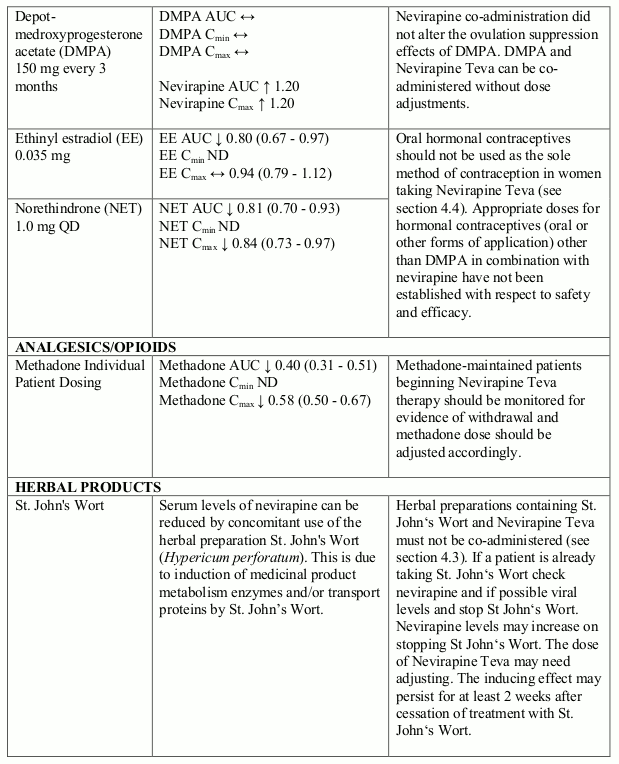
Hormonal methods of birth control other than Depot-medroxyprogesterone acetate (DMPA) should not be used as the sole method of contraception in women taking Nevirapine Teva, since nevirapine might lower the plasma concentrations of these medicinal products. For this reason, and to reduce the risk of HIV transmission, barrier contraception (e.g. condoms) is recommended. Additionally, when postmenopausal hormone therapy is used during administration of nevirapine, its therapeutic effect should be monitored.
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
In clinical studies, nevirapine has been associated with an increase in HDL-cholesterol and an overall improvement in the total to HDL-cholesterol ratio. However, in the absence of specific studies, the clinical impact of these findings is not known. In addition, nevirapine has not been shown to cause glucose disturbances.
Osteonecrosis
Although the etiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Immune Reactivation Syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
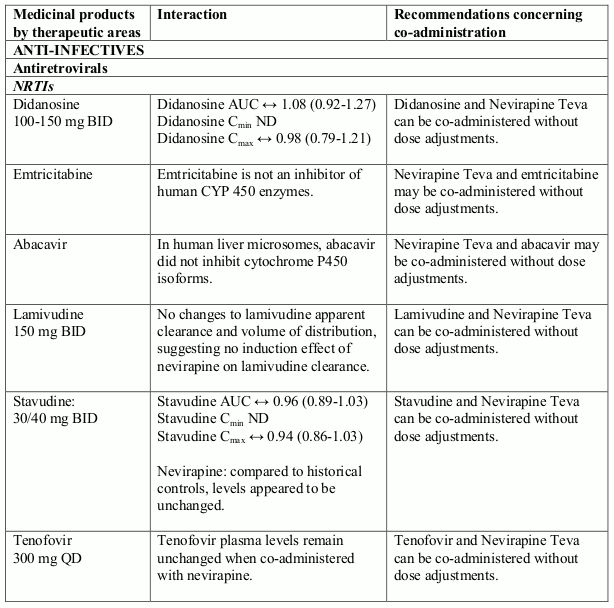
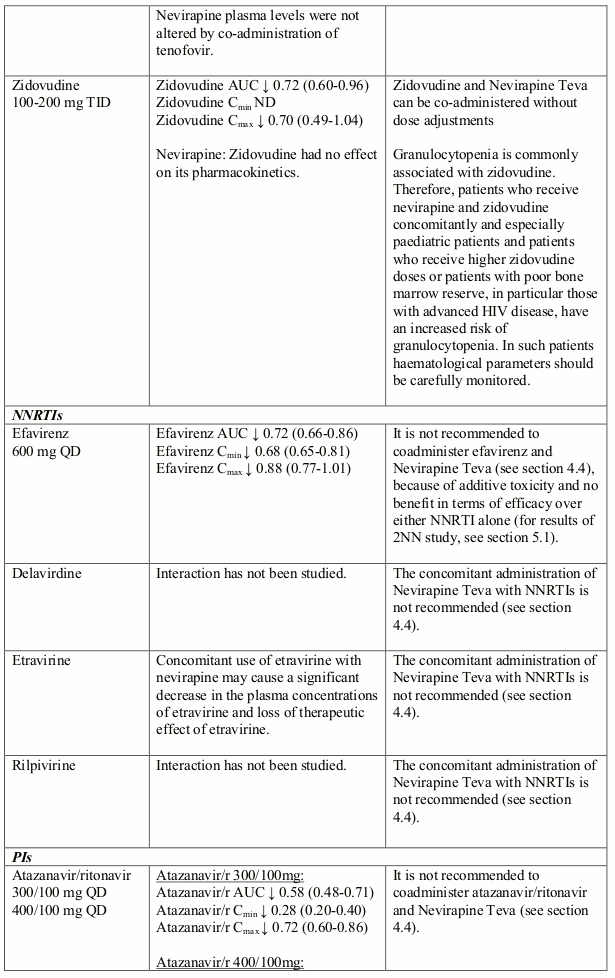
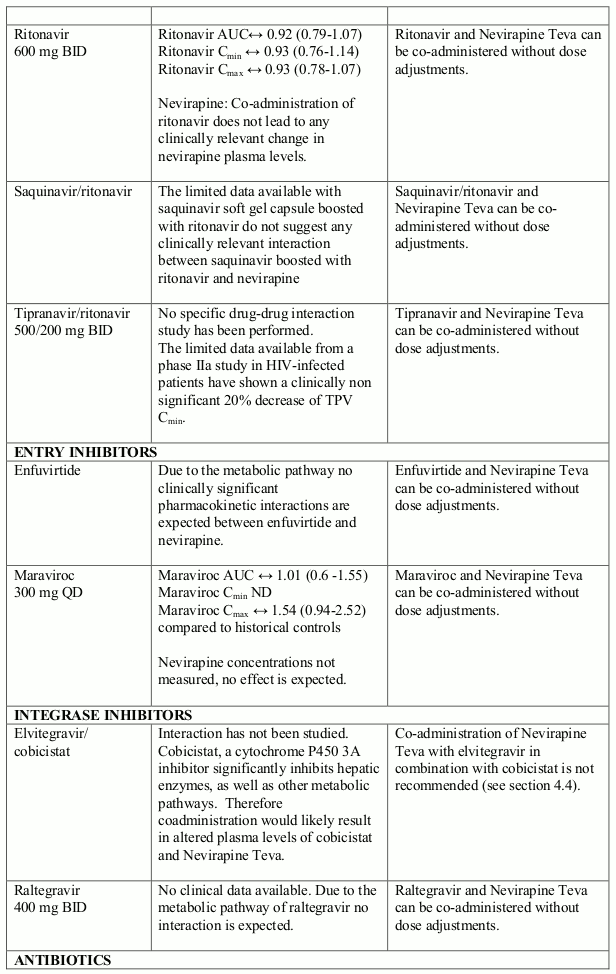
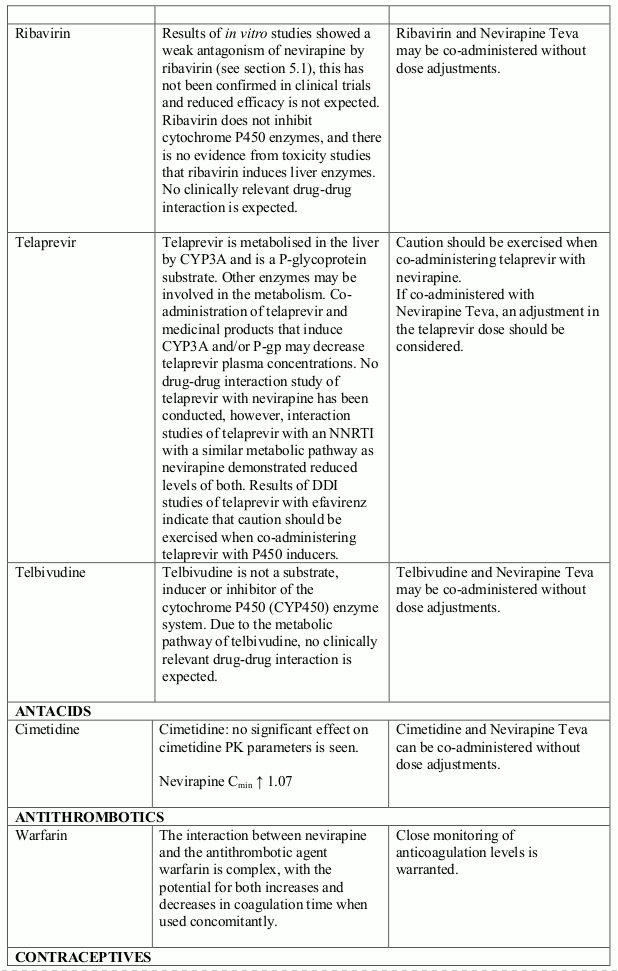
The available pharmacokinetic data suggest that the concomitant use of rifampicin and nevirapine is not recommended. Furthermore, combining the following compounds with Nevirapine Teva is not recommended: efavirenz, ketoconazole, delavirdine, etravirine, rilpivirine, elvitegravir (in combination with cobicistat), atazanavir (in combination with ritonavir), boceprevir; fosamprenavir (if not co-administered with low dose ritonavir) (see section 4.5).
Granulocytopenia is commonly associated with zidovudine. Therefore, patients who receive nevirapine and zidovudine concomitantly and especially paediatric patients and patients who receive higher zidovudine doses or patients with poor bone marrow reserve, in particular those with advanced HIV disease, have an increased risk of granulocytopenia. In such patients haematological parameters should be carefully monitored.
Lactose
Nevirapine Teva tablets contain 336 mg of lactose per maximum recommended daily dose. Patients with rare hereditary problems of galactose intolerance e.g. galactosaemia, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Interaction with other medicinal products and other forms of interaction
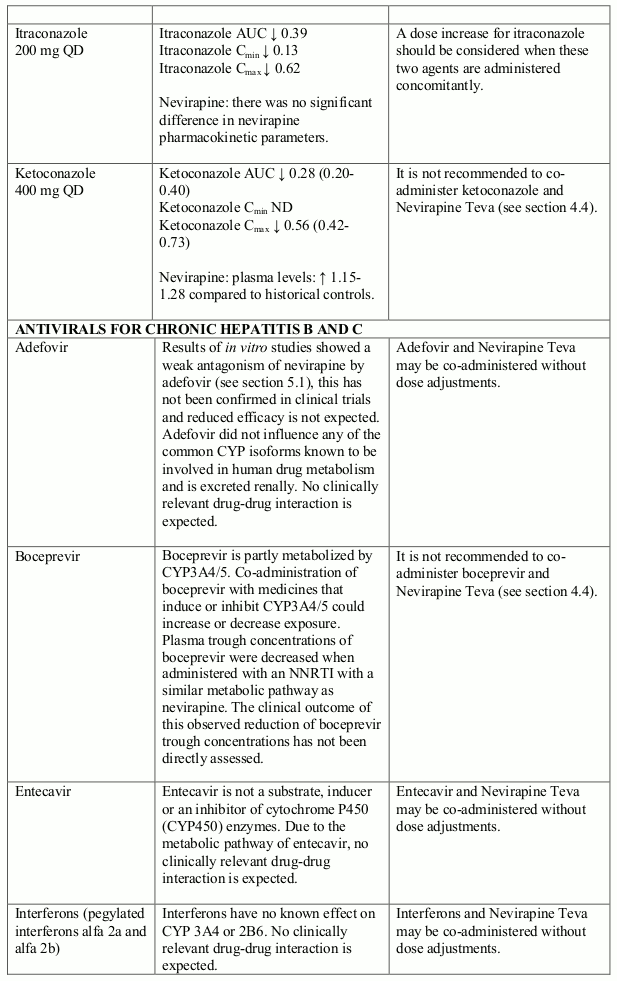
Nevirapine is an inducer of CYP3A and potentially CYP2B6, with maximal induction occurring within 2-4 weeks of initiating multiple-dose therapy.
Compounds using this metabolic pathway may have decreased plasma concentrations when co- administered with nevirapine. Careful monitoring of the therapeutic effectiveness of P450 metabolised medicinal products is recommended when taken in combination with nevirapine.
The absorption of nevirapine is not affected by food, antacids or medicinal products which are formulated with an alkaline buffering agent.
The interaction data is presented as geometric mean value with 90% confidence interval (90% CI) whenever these data were available. ND = Not Determined, ↑ = Increased, ↓ = Decreased, ↔ = No Effect


Other information
Nevirapine metabolites
Studies using human liver microsomes indicated that the formation of nevirapine hydroxylated metabolites was not affected by the presence of dapsone, rifabutin, rifampicin, and trimethoprim/sulfamethoxazole. Ketoconazole and erythromycin significantly inhibited the formation of nevirapine hydroxylated metabolites.
Fertility, pregnancy and lactation
Women of childbearing potential / Contraception in males and females
Women of childbearing potential should not use oral contraceptives as the sole method for birth control, since nevirapine might lower the plasma concentrations of these medicinal products (see sections 4.4 & 4.5).
Pregnancy
Currently available data on pregnant women indicate no malformative or foeto/neonatal toxicity. To date no other relevant epidemiological data are available. No observable teratogenicity was detected in reproductive studies performed in pregnant rats and rabbits (see section 5.3). There are no adequate and well-controlled studies in pregnant women. Caution should be exercised when prescribing nevirapine to pregnant women (see section 4.4). As hepatotoxicity is more frequent in women with CD4 cell counts above 250 cells/mm³ with detectable HIV-1 RNA in plasma (50 or more copies/ml), these conditions should be taken in consideration on therapeutic decision (see section 4.4). There is not enough evidence to substantiate that the absence of an increased risk for toxicity seen in pretreated women initiating nevirapine with an undetectable viral load (less than 50 copies/ml of HIV-1 in plasma) and CD4 cell counts above 250 cells/mm³ also applies to pregnant women. All the randomised studies addressing this issue specifically excluded pregnant women, and pregnant women were under-represented in cohort studies as well as in meta-analyses.
Breast-feeding
Nevirapine readily crosses the placenta and is found in breast milk.
It is recommended that HIV-infected mothers do not breast-feed their infants to avoid risking postnatal transmission of HIV and that mothers should discontinue breast-feeding if they are receiving nevirapine.
Fertility
In reproductive toxicology studies, evidence of impaired fertility was seen in rats.
Effects on ability to drive and use machines
There are no specific studies about the ability to drive vehicles and use machinery. However, patients should be advised that they may experience adverse reactions such as fatigue during treatment with Nevirapine Teva. Therefore, caution should be recommended when driving a car or operating machinery. If patients experience fatigue they should avoid potentially hazardous tasks such as driving or operating machinery.
Undesirable effects
Summary of the safety profile
The most frequently reported adverse reactions related to nevirapine therapy, across all clinical studies, were rash, allergic reactions, hepatitis, abnormal liver function tests, nausea, vomiting, diarrhoea, abdominal pain, fatigue, fever, headache and myalgia.
The postmarketing experience has shown that the most serious adverse reactions are Stevens-Johnson syndrome/toxic epidermal necrolysis, serious hepatitis/hepatic failure, and drug reaction with eosinophilia and systemic symptoms, characterised by rash with constitutional symptoms such as fever, arthralgia, myalgia and lymphadenopathy, plus visceral involvement, such as hepatitis, eosinophilia, granulocytopenia, and renal dysfunction. The first 18 weeks of treatment is a critical period which requires close monitoring (see section 4.4).
Tabulated summary of adverse reactions
The following adverse reactions which may be causally related to the administration of nevirapine have been reported. The frequencies estimated are based on pooled clinical study data for adverse reactions considered related to nevirapine treatment. Frequency is defined using the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000).
Blood and lymphatic system disorders
Common: granulocytopenia
Uncommon: anaemia
Immune system disorders
Common: hypersensitivity (incl. anaphylactic reaction, angioedema, urticaria)
Uncommon: anaphylactic reaction
Rare: drug reaction with eosinophilia and systemic symptoms
Nervous system disorders
Common: headache
Gastrointestinal disorders
Common: nausea, vomiting, abdominal pain, diarrhoea
Hepatobiliary disorders
Common: hepatitis (including severe and life-threatening hepatotoxicity) (1.9%)
Uncommon: jaundice
Rare: hepatitis fulminant (which may be fatal)
Skin and subcutaneous tissue disorders
Very common: rash (12.5%)
Uncommon: Stevens-Johnson syndrome/toxic epidermal necrolysis (which may be fatal) (0.2%), angioedema, urticaria
Musculoskeletal and connective tissue disorders
Uncommon: arthralgia, myalgia
General disorders and administration site conditions
Common: pyrexia, fatigue
Investigations
Common: liver function tests abnormal (alanine aminotransferase increased; transaminases increased; aspartate aminotransferase increased; gamma-glutamyltransferase increased; hepatic enzyme increased; hypertransaminasaemia)
Uncommon: blood phosphorus decreased; blood pressure increased
Description of selected adverse reactions
In study 1100.1090, from which the majority of related adverse events (n=28) were received, patients on placebo had a higher incidence of events of granulocytopenia (3.3%) than patients on nevirapine (2.5%).
Anaphylactic reaction was identified through post-marketing surveillance but not observed in randomised, controlled clinical studies. The frequency category was estimated from a statistical calculation based on the total number of patients exposed to nevirapine in randomised controlled clinical studies (n=2,718).
Decreased blood phosphorus and increased blood pressure were observed in clinical studies with coadministration of tenofovir/emtricitabine.
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
The following adverse reactions have also been reported when nevirapine has been used in combination with other anti-retroviral agents: pancreatitis, peripheral neuropathy and thrombocytopaenia. These adverse reactions are commonly associated with other antiretroviral agents and may be expected to occur when nevirapine is used in combination with other agents; however it is unlikely that these adverse reactions are due to nevirapine treatment. Hepatic-renal failure syndromes have been reported rarely.
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
Skin and subcutaneous tissues
The most common clinical toxicity of nevirapine is rash, with nevirapine attributable rash occurring in 12.5% of patients in combination regimens in controlled studies.
Rashes are usually mild to moderate, maculopapular erythematous cutaneous eruptions, with or without pruritus, located on the trunk, face and extremities. Hypersensitivity (anaphylactic reaction, angioedema and urticaria) have been reported. Rashes occur alone or in the context of drug reaction with eosinophilia and systemic symptoms, characterised by rash with constitutional symptoms such as fever, arthralgia, myalgia and lympadenopathy, plus visceral involvement, such as hepatitis, eosinophilia, granulocytopenia, and renal dysfunction.
Severe and life-threatening skin reactions have occurred in patients treated with nevirapine, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Fatal cases of SJS, TEN and drug reaction with eosinophilia and systemic symptoms have been reported. The majority of severe rashes occurred within the first 6 weeks of treatment and some required hospitalisation, with one patient requiring surgical intervention (see section 4.4).
Hepato-biliary
The most frequently observed laboratory test abnormalities are elevations in liver function tests (LFTs), including ALAT, ASAT, GGT, total bilirubin and alkaline phosphatase. Asymptomatic elevations of GGT levels are the most frequent. Cases of jaundice have been reported. Cases of hepatitis (severe and life-threatening hepatotoxicity, including fatal fulminant hepatitis) have been reported in patients treated with nevirapine. The best predictor of a serious hepatic event was elevated baseline liver function tests. The first 18 weeks of treatment is a critical period which requires close monitoring (see section 4.4).
Paediatric population
Based on clinical study experience of 361 paediatric patients the majority of which received combination treatment with ZDV or/and ddI, the most frequently reported adverse events related to nevirapine were similar to those observed in adults. Granulocytopenia was more frequently observed in children. In an open-label clinical study (ACTG 180) granulocytopenia assessed as medicinal product-related occurred in 5/37 (13.5%) of patients. In ACTG 245, a double-blind placebo controlled study, the frequency of serious medicinal product-related granulocytopenia was 5/305 (1.6%). Isolated cases of Stevens-Johnson syndrome or Stevens-Johnson/toxic epidermal necrolysis transition syndrome have been reported in this population.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.