NOCDURNA Orally disintegrating tablet Ref.[27533] Active ingredients: Desmopressin
Source: Health Products and Food Branch (CA) Revision Year: 2014
Action and clinical pharmacology
Mechanism of Action
The antidiuretic effects of desmopressin are mediated by stimulation of V2-receptors thereby increasing water re-absorption in the kidney, and hence reducing urine production. Stimulation of V2-receptors may also cause an increase in the levels of blood coagulation factors, factor VIII and von Willebrand factor, but this effect occurs at higher doses of desmopressin than those required for inducing antidiuresis.42
Pharmacodynamics
NOCDURNA contains desmopressin, a structural analogue of the natural pituitary hormone arginine vasopressin. The difference lies in the desamination of cysteine and substitution of Larginine by D-arginine. This results in a considerably longer duration of action and a complete lack of pressor effect in the dosages clinically used.
In study CS29, the weight-corrected NOCDURNA dose that induced 50% maximum achievable drug effect on nocturnal urine volume differed significantly between females and males. The estimated exposure for males was 2.7 fold (95% CI: 1.3-8.1) higher than the value for females to obtain an identical dynamic effect, corresponding to higher desmopressin sensitivity among females.3
Gender and age appear to have a dose-dependent effect on hyponatremia. The incidences of hyponatremia rise with increasing dose and with increasing age.
Pharmacokinetics
Absorption
The overall mean absolute bioavailability of desmopressin orally disintegrating tablets administered sublingually from earlier dose-ranging studies of doses of 200, 400 and 800 is 0.25% with a 95% confidence interval of 0.21–0.31%. 9 Desmopressin exhibits a moderate-to-high variability in bioavailability, both within and between subjects. 10 Desmopressin orally disintegrating tablets show dose linearity regarding AUC and Cmax in the range of 60 to 240.7 However, the bioavailability of doses below 60 has not been evaluated.
Distribution
The distribution volume of desmopressin after intravenous administration is 33 L.
Metabolism
Desmopressin did not show any effect on any of the nine CYP 450 subtypes. In vivo drug-drug interactions based on activation or inhibition of CYP P450 are therefore very unlikely.-20
Excretion
Desmopressin is mainly excreted in the urine. After intravenous injection, 52% of the dose could be recovered in the urine within 24 hours as unchanged desmopressin. The geometric mean terminal half-life is 2.8 (CV=24%) hours.8
Terminal half-life significantly increases in patients with severe renal impairment (see Warnings and Precautions).8
Special Populations and Conditions
Geriatric
The pharmacokinetics of desmopressin acetate in the nocturia population does not differ from those in healthy subjects. A pooled data analysis demonstrated no significant correlation between age and pharmacokinetics, and no gender-related difference in AUCinf could be found. In very elderly patients, a decrease in the renal elimination of desmopressin could be expected. 34
Use of NOCDURNA requires careful fluid restriction to prevent possible prolonged fluid retention and subsequent hyponatremia. Fluid restriction should be discussed with the patient [See Warnings and Precautions and Gender and Age Effects].
Gender and Age Effect
A two part study involving single oral dosage with 400 µg desmopressin (Part A) and a randomized, placebo-controlled, 2 way crossover evaluation involving treatment with 400 µg desmopressin or placebo for 3 consecutive nights with a 7-14 day washout period showed that the pharmacokinetic parameters in elderly (>65 years) nocturic subjects did not differ from those in healthy subjects. A gender-related difference in the extent but not in the rate of absorption was observed. 11 A subsequent pooled data analysis of several clinical studies did not confirm this difference. The linear relationship between plasma desmopressin concentrations at 2-3 hours post-dose and AUCinf in these subjects suggests that plasma desmopressin concentrations at 2 (or 3) hours can be used as an accurate predictor of AUCinf in elderly subjects with nocturia.
The low level of absorption of desmopressin following oral administration means that a high level of variability may be expected. A study evaluating intra and inter individual variation in pharmacokinetics of desmopressin after three administration in an oral lyophilisate to healthy nonsmoking volunteers showed no gender differences in intra and inter-subject variability in AUC-∞ AUC, or Cmax. After multiple (3) doses of desmopressin Melt at 200 µg, the PK parameters did not differ statistically significantly after each dose.10
The large Phase 3 study with desmopressin Melt CS029 is the first to show a clear gender difference in the effect of desmopressin on nocturnal urine volume. A further analysis was performed on the data obtained from nocturia patients in this study, together with single-dose pharmacokinetic data from healthy subjects in a three-period crossover study comparing the exposure of oral lyophilisate containing 60, 120 and 240 µg desmopressin in 24 non-smoking subjects6 and an open-labeled, randomized, two period crossover study investigating the relative bioavailability of two single doses of the currently marketed MINIRIN Tablets (2 x 200 µg) and a single dose of desmopressin administered as a new orodispersible tablet (240 µg).6
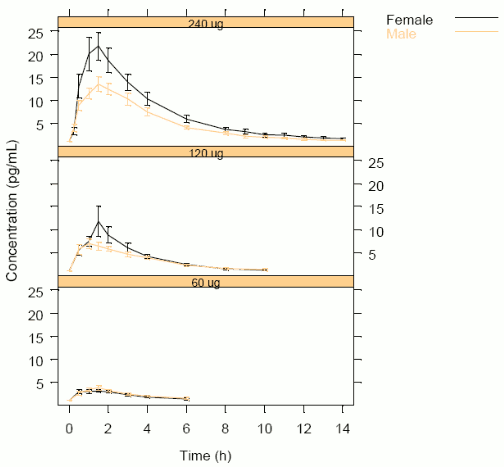
Mean desmopressin concentration profiles are shown by dose and gender in Figure 1. Age and gender were found not to be statistically significant while weight was significant with respect to log(Cmax) and borderline significant with respect to log(AUC). Gender differences in drug exposure were not statistically significant when adjusting for body weight.
Figure 1. Modelling Analysis: Mean Desmopressin Concentration by Dose and Gender:
The incidence of hyponatremia increased with increasing doses of NOCDURNA. In CS29 and CS31, hyponatremia referred to serum sodium below 130 mmol/L, and patients continued treatment unless serum sodium fell below 125 mmol/L. No female subject who received NOCDURNA 25 µg had serum sodium levels below 125 mmol/L and none of these subjects discontinued therapy due to hyponatremia. No males under 65 years of age had serum sodium levels below 125 mmol/L. During CS29, among males ≥65 years of age, 2 who received NOCDURNA 100 µg and 1 who received 50 µg NOCDURNA had serum sodium levels fall below 125 mmol/L; all 3 subjects were discontinued, 2 due to "hyponatremia" and 1 due to "blood sodium decreased." Minimum post baseline serum sodium levels by age groups in females treated with NOCDURNA 25 µg and males treated with NOCDURNA 50 µg is presented by age group for CS29 and CS31 in Table 4.3,4
Table 4. Minimum Post-Baseline Serum Sodium Levels by Age Group in Females Treated with NOCDURNA 25 µg and Males treated with NOCDURNA 50µg (Overall Safety population CS29+CS31):
| Serum Sodium (mmol/L) | NOCDURNA 25 µg for Females | NOCDURNA 50 µg for Males | ||
|---|---|---|---|---|
| <65 years (N=59) | ≥65 years (N=37) | <65 years (N=53) | ≥65 years (N=65) | |
| Observed N* | 59 (100%) | 37 (100%) | 52 (98.1%) | 65 (100%) |
| ≥135 | 48 (81.4%) | 27 (73.0%) | 37 (69.8%) | 33 (50.8%) |
| 130-134 | 10 (16.9%) | 6 (16.2%) | 15 (28.3%) | 21 (32.3%) |
| 125-129 | 1 (1.7%) | 4 (10.8%) | 0 | 10 (15.4%) |
| <125 | 0 | 0 | 0 | 1 (1.5%) |
Cross reference: [Table 14.8.1.9 ]CS31 CTR – Overall safety CS29 and CS31
* Number of subjects with data
CS41 Part I
During CS41 Part I, 4 males who received NOCDURNA 75 µg and 2 who received NOCDURNA 50 µg had serum sodium less than or equal to 125 mmol/L; all 6 subjects were discontinued, 5 due to "hyponatremia" and 1 due to "dizziness". No males <65 years of age who received NOCDURNA 50 µg and one male who received NOCDURNA 75 µg had serum sodium ≤125 mmol/L. Minimum post baseline serum sodium levels in males who received NOCDURNA 50 µg is presented by age group for CS41 Part I in Table 5.5
Table 5. Minimum Post-Baseline Serum Sodium Levels by Age Group in Males Treated with NOCDURNA 50µg (CS41 Part I):
| Serum Sodium (mmol/L) | NOCDURNA 50 µg (N=119) | |
|---|---|---|
| Age <65 (N=62) | Age ≥65 (N=57) | |
| ≥135 | 60 (97%) | 48 (84%) |
| 130-134 | 2 (3%) | 7 (12%) |
| 126-129 | 0 | 0 |
| ≤125 | 0 | 2 (4%) |
Cross reference: [Table 7.1.1.5 and 7.1.1.6] CS41 CTR
Due to the rare cases of serum sodium levels ≤125 mmol/L in males ≥65 years who received NOCDURNA 50 µg in the Phase 3 trials, serum monitoring is warranted in this group.3,4,5
Toxicology
(i) Acute Toxicity
The i.v. acute toxicity of desmopressin acetate was studied in mice, rats and rabbits. Mice tolerated i.v. doses of 2 mg/kg. At doses of 30 µg/kg in rats and 50 µg/kg in rabbits, only transient changes in clinical behaviour were observed.12,13,41
(ii) Subacute Toxicity
Results from 14-day studies show that the drug given intravenously to rats at 8 µg/kg/day and to rabbits at 6 µg/kg/day caused no biologically significant changes in hematological and clinical chemistry parameters. Post-mortem examinations did not reveal any abnormalities.14,15
Rats which received 5 mg/kg/day subcutaneously for 3 days did not show any significant changes in weight, blood count, or organ changes.41
(iii) Chronic Toxicity
Subcutaneous Administration
Rat Studies:
In a controlled 8-week experiment, 20 rats received 2 µg/kg/day desmopressin acetate subcutaneously. No increase in blood glucose or morphological or histological pancreatic changes occurred.42
Rats (20 per group) which received doses of 5, 50 and 500 ng/kg/day, for six months did not show any significant changes in weight, blood values, or levels of transaminases. The weight of heart, lungs and kidneys decreased in female animals in the lower dose groups but not in the higher ones. In the male animals a decrease in non-esterified fatty acids was noted.41
Rats were treated with dose levels of 0.1, 1 and 10 µg/kg/day by subcutaneous injection for 13 weeks. Anticipated pharmacological changes were observed in a series of urinary parameters mainly at the high dose: decreased urinary volume and pH; increased specific gravity and concentration of sodium, potassium, chloride and protein. Absolute and relative kidney weights were increased at all dose levels. There were no macroscopic observations at necropsy. Histopathology showed changes at the injection site, where the incidence and severity were slightly increased at the high dose level.
Dog Studies:
Dogs (3 per group) which received subcutaneous doses of 10 and 100 ng/kg/day for 6 months did not show any significant changes in comparison with control groups in blood sugar or transaminases and did not show histological or morphological organ changes.41
Intravenous Administration
Rat Studies:
In rats treated intravenously with desmopressin at dose levels of 9.47, 47.4 or 238 μg/kg/day for 180 days, urinary specific gravity was increased and urinary volume was decreased in all desmopressin treated groups, thus showing the pharmacological effect of the compound. Absolute kidney weight was increased from the mid dose level while body weight related kidney weight was increased from the low dose level. There were no gross pathological changes at necropsy. Histopathological changes were confined to the kidneys and consisted of increased incidence of tubular protein casts from 47.4 μg/kg/day and hyaline droplet degeneration at the high dose level. All changes were reversible after the 30-day reversibility period, except increased kidney weight, which showed incomplete reversibility. 15
Oral Administration
Rat Studies:
Oral administration of desmopressin to rats (20 male and 20 females per group dosed at 25, 75 and 200 µg/kg/day) did not reveal any clinical findings related to desmopressin. Treated male and female rats were comparable to controls with respect to food consumption, body weight gain and water consumption. There were no drug-induced ocular abnormalities.21
A dosage-related reduction was seen in levels of total circulating white blood cells, attributable to reduced neutrophil and lymphocyte counts in treated females, when compared with controls, at the week 13 and 26 investigations. Treated males were not affected. 21Reduced plasma Factor VIII levels were seen in treated females at week 14 and treated males at week 25 in comparison with controls.21
The terminal studies revealed no morphological or histological changes related to treatment with desmopressin.21
Dog Studies:
When desmopressin was given orally to dogs (4 males and 4 females per group, at 0, 25, 75 and 200 µg/kg/day) all animals survived the 26-week period and no clinical signs were observed that were related to treatment. There were no adverse effects on body weight, food and water consumption and no ocular abnormalities. Hematological investigations revealed no treatmentrelated findings.16
During weeks 6, 13 and 26 serum total protein concentrations of treated animals were increased due to an increase in the globulin fraction. However, there were no changes from the pre-dose values in males at 200 µg/kg/day after 13 and 26 weeks treatment and males at 75 µg/kg/day after 26 weeks treatment.16
No organ morphological or histological changes were seen on autopsy which could be related to treatment with desmopressin.16
Reproduction Studies
Subcutaneous Administration
Rat Studies:
In a teratogenicity study in Wistar rats, neither teratologic nor embryotoxic effects were observed in 369 foetuses from 40 females dosed with up to 50 ng/kg/day desmopressin acetate subcutaneously during day 1 to day 20 of gestation.40
Rabbit Studies:
In a study of 78 Dutch belted rabbits which received subcutaneous doses of desmopressin acetate up to 10 µg/kg/day during day 6 and day 18 of pregnancy, neither teratogenic nor embryotoxic effects were observed in 296 fetuses. Weaning was unaffected.39
Intravenous Administration
Rat Studies:
A teratology study was performed in rats. Groups of 30 pregnant Slc:Wistar rats were treated daily from day 7 to day 17 of gestation by i.v. administration of desmopressin at dosage levels of 9.47, 47.4 and 238 g desmopressin/kg/day. A control group received the vehicle, physiological saline. Twenty females in each group were killed on day 20 of gestation to allow fetal examinations; the remaining 10 females were allowed to litter to determine any postnatal effects that might be attributable to prenatal treatment. There were no effects of treatment on the dams, and fetal survival, growth and morphology were also unaffected. Postnatal offspring survival, growth, development, behavior and reproductive performance also showed no effects of prenatal exposure to desmopressin.22
Genotoxicity
The genotoxic potential of desmopressin was examined in 3 Ames tests and one mouse lymphoma assay all of which turned out to be negative. Desmopressin is therefore considered to be devoid of mutagenic potential under the condition tested.22
Carcinogenesis
Studies with desmopressin acetate have not been performed to evaluate carcinogenic potential.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.