NUBEQA Film-coated tablet Ref.[9925] Active ingredients: Darolutamide
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Bayer AG, 51368, Leverkusen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Endocrine therapy, anti-androgens
ATC code: L02BB06
Mechanism of action
Darolutamide is an androgen receptor (AR) inhibitor with a flexible polar-substituted pyrazole structure that binds with high affinity directly to the receptor ligand binding domain. Darolutamide competitively inhibits androgen binding, AR nuclear translocation, and AR mediated transcription. A major metabolite, keto-darolutamide, exhibited similar in vitro activity to darolutamide. Darolutamide treatment decreases prostate tumour cell proliferation leading to potent antitumour activity.
Pharmacodynamic effects
No prolongation of the mean QTcF interval (i.e., greater than 10 ms) was observed after oral administration of 600 mg darolutamide twice daily compared to placebo.
Clinical efficacy and safety
Efficacy and safety were established in two randomised placebo-controlled multicentre phase III studies in patients with nmCRPC (ARAMIS) and mHSPC (ARASENS). All patients received a luteinising hormone-releasing hormone (LHRH) analogue concurrently or had a bilateral orchiectomy.
non-metastatic castration resistant prostate cancer (nmCRPC)
The efficacy and safety of darolutamide was assessed in a randomised, double-blind, placebo-controlled multicentre phase III study (ARAMIS) in patients with non-metastatic (as assessed by conventional imaging CT, bone scan, MRI) castration resistant prostate cancer with a prostate-specific antigen doubling time (PSADT) of ≤10 months.
Patients were included in the trial if they had 3 rising prostate-specific antigen (PSA) levels after the nadir taken at least 1 week apart during androgen deprivation therapy, PSA ≥2 ng/mL at screening and castrate level of serum testosterone <1.7 nmol/L.
Patients with a medical history of seizure were allowed to enter the study. There were 12 patients (0.21%) enrolled on the darolutamide arm with a history of seizure. Patients with uncontrolled hypertension or recent (in the past 6 months) stroke, myocardial infarction, severe/unstable angina pectoris, coronary/peripheral artery bypass graft, congestive heart failure New York Heart Association (NYHA) Class III or IV were excluded from the study.
Patients with prior treatment with second generation AR inhibitors such as enzalutamide, apalutamide and darolutamide, or CYP17 enzyme inhibitors such as abiraterone acetate as well as patients receiving systemic corticosteroid with dose greater than the equivalent 10 mg of prednisone/day within 28 days before randomisation were excluded from the study. In total, 1509 patients were randomized 2:1 to receive either 600 mg darolutamide orally twice daily (n=955) or matching placebo (n=554).
Patients with presence of pelvic lymph nodes <2 cm in short axis below the aortic bifurcation were allowed to enter the study. Absence or presence of metastasis was assessed by independent central radiological review. Included in these analyses were 89 patients that were retrospectively identified with metastasis at baseline. Randomization was stratified by PSADT (≤6 months or >6 months) and use of osteoclast-targeted therapy at study entry (yes or no).
The following patient demographics and disease characteristics were balanced between treatment arms. The median age was 74 years (range 48-95) and 9% of patients were 85 years of age or older. The racial distribution was 79% White, 13% Asian, and 3% Black. A majority of patients had a Gleason score of 7 or higher at diagnosis (73%). The median PSADT was 4.5 months. Nine percent (9%) of patients had prior orchiectomy, 25% of patients had prior prostatectomy and 50% of patients had at least one prior radiotherapy. Seventy-six percent (76%) of patients received more than one prior anti-hormonal treatment. Patients had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 (69%) or 1 (31%) at study entry.
Treatment with darolutamide continued until radiographic disease progression as assessed by conventional imaging (CT, bone scan, MRI) by blinded central review, unacceptable toxicity or withdrawal.
The primary efficacy endpoint was metastasis free survival (MFS). Secondary endpoints were overall survival (OS), time to pain progression, time to initiation of first cytotoxic chemotherapy for prostate cancer, and time to first symptomatic skeletal events (defined as occurrence of any of the following: external beam radiotherapy to relieve skeletal symptoms, new symptomatic pathologic bone fracture, spinal cord compression, or tumour-related orthopaedic surgical intervention).
Treatment with darolutamide resulted in an improvement in MFS compared to placebo (see Table 3 and Figure 1).
MFS results were consistent across patient subgroups regardless of PSADT, prior use of bone-targeting agents or loco-regional disease. Additional subgroups with consistent MFS results included PSA at baseline, Gleason score at diagnosis, age, geographical region, ECOG PS at baseline, race, and number of prior hormonal therapies.
After the primary analysis of MFS, once the study was unblinded, patients receiving placebo were offered treatment with open-label darolutamide (cross-over option). Among the 554 patients randomised to placebo, 170 (31%) crossed over to receive darolutamide treatment. The OS analysis was not adjusted for confounding effects of cross-over.
At the time of the final analysis, treatment with darolutamide resulted in a statistically significant improvement in overall survival compared to placebo (median was not reached in either arm, see Table 3 and Figure 2).
Treatment with darolutamide also resulted in statistically significant delays in time to pain progression, time to initiation of first cytotoxic chemotherapy and time to first symptomatic skeletal event compared to placebo (see Table 3).
At the time of final analysis, the median duration of treatment in patients treated with darolutamide was 33.3 months (range: 0.0 to 74.0 months) during combined double-blind and open-label period.
All analyses were performed in the full analysis set.
Table 3. Efficacy results from the ARAMIS study:
| Efficacy parameter | Number (%) of patients with events | Median (months) (95% CI) | Hazard Ratiob (95% Confidence Interval [CI]) p-value (two-sided) | ||
|---|---|---|---|---|---|
| Darolutamide (N=955) | Placeboa (N=554) | Darolutamide (N=955) | Placeboa (N=554) | ||
| Metastasis free survivalc | 221 (23.1%) | 216 (39.0%) | 40.4 (34.3, NR) | 18.4 (15.5, 22.3) | 0.413 (0.341, 0.500) <0.000001 |
| Overall survival | 148 (15.5%) | 106 (19.1%) | NR (56.1, NR) | NR (46.9, NR) | 0.685 (0.533, 0.881) 0.003048 |
| Time to pain progressionc,d | 251 (26.3%) | 178 (32.1%) | 40.3 (33.2, 41.2) | 25.4 (19.1, 29.6) | 0.647 (0.533, 0.785) 0.000008 |
| Time to initiation of first cytotoxic chemotherapy | 127 (13.3%) | 98 (17.7%) | NR (NR, NR) | NR (NR, NR) | 0.579 (0.444, 0.755) 0.000044 |
| Time to first symptomatic skeletal event | 29 (3.0%) | 28 (5.1%) | NR (NR, NR) | NR (NR, NR) | 0.484 (0.287, 0.815) 0.005294 |
a including 170 patients who crossed over to open-label darolutamide
b Hazard ratio <1 favours darolutamide
c for MFS and time to pain progression, the analysis performed at the time of primary completion is considered as the final analysis
d Patient reported outcome as evaluated by Brief Pain Inventory-Short Form questionnaire
NR: Not reached.
Treatment with darolutamide resulted in a longer progression free survival (PFS, median 36.8 vs 14.8 months, HR=0.380, nominal p<0.000001) and time to PSA progression (median 29.5 vs 7.2 months, HR=0.164, nominal p<0.000001). Consistency of effect was observed across all measures of survival (MFS, OS and PFS).
Figure 1. Kaplan-Meier curves of metastasis free survival (ARAMIS):

Figure 2. Kaplan-Meier curves of overall survival (ARAMIS):

Patients receiving darolutamide in the ARAMIS study (double-blind period) demonstrated a significantly higher confirmed PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 84.0% vs 7.9% (difference = 76.1%, p<0.000001 (nominal p-value, for information only)).
metastatic hormone-sensitive prostate cancer (mHSPC)
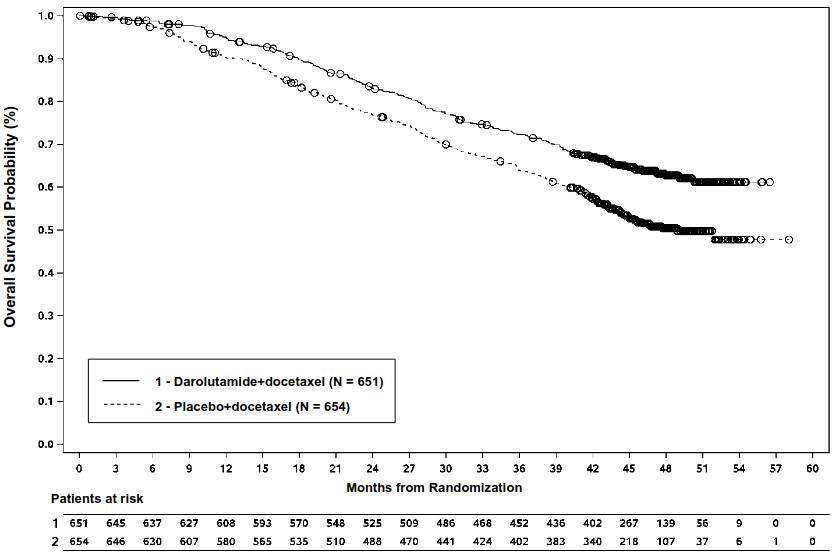
The efficacy and safety of darolutamide in combination with docetaxel was assessed in a multicentre, double-blind, placebo-controlled phase III study (ARASENS) in patients with mHSPC. In total, 1306 patients were randomised 1:1 to receive 600 mg darolutamide orally twice daily (n=651) or matching placebo (n=655), concomitantly with 75 mg/m² of docetaxel for 6 cycles. Treatment with darolutamide or placebo continued until symptomatic progressive disease, change of antineoplastic therapy, unacceptable toxicity, death, or withdrawal.
Presence of metastasis was assessed by independent central radiological review. Patients with regional lymph node involvement only (M0) were excluded from the study. Randomisation was stratified by extent of disease (non-regional lymph nodes metastases only (M1a), bone metastases with or without lymph node metastases (M1b) or visceral metastases with or without lymph node metastases or with or without bone metastases (M1c)) and by alkaline phosphatase level (< or ≥ upper limit of normal) at study entry. Patients with brain metastases were allowed to enter the study but there were no patients with brain metastasis enrolled.
The following patient demographics and disease characteristics were balanced between treatment arms. The median age was 67 years (range 41-89) and 0.5% of patients were 85 years of age or older. The racial distribution was 52% White, 36% Asian, and 4% Black. A majority of patients had a Gleason score of 8 or higher at diagnosis (78%). 71% of patients had an ECOG PS score of 0 and 29% of patients had an ECOG PS score of 1. There were 86.1% of patients with de novo and 12.9% of patients with recurrent disease. At study entry 3% of patients had M1a, 79.5% had M1b and 17.5% had M1c; alkaline phosphatase was < ULN in 44.5% of patients and ≥ ULN in 55.5% of patients; median PSA level at baseline was 30.3 μg/L and 24.2 μg/L for darolutamide vs the placebo group, respectively. Patients with a medical history of seizure were allowed to enter the study, and 4 patients (0.6%) were enrolled in the darolutamide+docetaxel arm.
77.0% of patients had high-volume disease and 23.0% had low-volume disease. High-volume disease was defined as presence of visceral metastases or 4 or more bone lesions, with at least 1 metastasis beyond the vertebral column and pelvic bones. Around 25% of patients received concomitant treatment with bisphosphonates or denosumab.
The primary efficacy endpoint was overall survival (OS). Secondary endpoints were time to castration-resistant prostate cancer, time to pain progression, symptomatic skeletal event free survival (SSE-FS), time to first symptomatic skeletal event (SSE), time to initiation of subsequent antineoplastic therapy, time to worsening of disease-related physical symptoms, and time to initiation of opioid use for ≥ 7 consecutive days. Pain progression was assessed using the patient-reported outcome (PROs) Brief Pain Inventory-Short Form (BPI-SF), defined as at least a 2-point worsening from nadir, and initiation of short- or long-acting opioid use for pain for ≥7 consecutive days.
The median duration of treatment was 41.0 months (range: 0.1 to 56.5 months) in patients treated with darolutamide+docetaxel and 16.7 months (range: 0.3 to 55.8 months) in patients treated with placebo+docetaxel. 87.6% and 85.5% of patients received full 6 cycles of docetaxel and 1.5% and 2.0% of patients did not receive docetaxel in darolutamide+docetaxel and placebo+docetaxel arm, respectively.
Table 4. Efficacy results from the ARASENS study:
| Efficacy parameter | Number (%) of patients with events | Median (months) (95% CI) | Hazard Ratiob (95% Confidence Interval [CI]) p-value (one-sided)c | ||
|---|---|---|---|---|---|
| Darolutamide + docetaxel (N=651) | Placebo + docetaxel (N=654)a | Darolutamide + docetaxel (N=651) | Placebo + docetaxel (N=654)a | ||
| Overall survivald | 229 (35.2%) | 304 (46.5%) | NR (NR, NR) | 48.9 (44.4, NR) | 0.675 (0.568, 0.801) <0.0001 |
a one patient in placebo arm was excluded from all analyses
b Hazard ratio <1 favours darolutamide
c based on stratified log-rank test
d OS results were consistent across patient subgroups, including extent of disease and alkaline phosphatase levels
NR: not reached
The following secondary efficacy endpoints showed a statistically significant advantage in favour of the patients in the darolutamide+docetaxel arm compared to patients in the placebo+docetaxel arm: time to castration-resistant prostate cancer (median NR vs 19.1 months; HR=0.357, p<0.0001); time to first symptomatic skeletal event (median NR vs NR months; HR=0.712, p=0.0081); time to initiation of subsequent antineoplastic chemotherapy (median NR vs 25.3 months; HR=0.388, p<0.0001); time to pain progression (median NR vs 27.5 months; HR=0.792, p=0.0058); symptomatic skeletal event free survival time (median 51.2 vs 39.7 months; HR=0.609, p<0.0001).
Figure 3. Kaplan-Meier curves of overall survival (ARASENS)a:
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with darolutamide in all subsets of the paediatric population in prostate malignant neoplasms (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
General introduction
Darolutamide consists of two diastereomers [(S,R)-darolutamide and (S,S)-darolutamide] which interconvert via the main circulating metabolite called keto-darolutamide. In vitro, all three substances show similar pharmacological activity. Darolutamide is poorly soluble in aqueous solvents over a large pH range and generally more soluble in organic solvents.
Absorption
Following oral administration of 600 mg (2 tablets of 300 mg) twice daily, peak plasma concentrations of darolutamide at steady state were 4.79 mg/L (coefficient of variation: 30.9%) in nmCRPC patients in the ARAMIS study and 3.84 mg/L (coefficient of variation: 35.6%) in mHSPC patients in the ARASENS study. Median time to achieve peak plasma concentrations was 3 to 4 hours. The ratio of the two diastereomers, (S,R)-darolutamide to (S,S)-darolutamide, changed from a 1:1 ratio in the tablet to an approximately 1:9 ratio in plasma based on AUC0-12 data at steady-state. Following oral administration together with food, steady-state is reached after 2-5 days of repeated twice-daily dosing.
The absolute bioavailability compared to an intravenous injection is approximately 30% following oral administration of a NUBEQA tablet containing 300 mg darolutamide under fasted conditions. Bioavailability of darolutamide was enhanced by 2.0- to 2.5-fold when administered with food. A similar increase of exposure was observed for the major metabolite keto-darolutamide.
Distribution
The apparent volume of distribution of darolutamide after intravenous administration is 119 L indicating that darolutamide is widely distributed throughout the body to both intracellular and extracellular fluid spaces.
Darolutamide is moderately (92%) bound to human plasma proteins without any difference between the two diastereomers. The major metabolite of darolutamide, keto-darolutamide, is highly (99.8%) bound to plasma proteins.
Passage of darolutamide across the blood-brain barrier has not been studied clinically. However, brain exposures to darolutamide in terms of AUC0-24 are very low with 4.5% of plasma exposure after single dose in rats and 1.9-3.9% after repeated dose in mice. This indicates low passage of darolutamide across the intact blood-brain barrier in rats and mice and a low likelihood that darolutamide crosses the intact blood-brain barrier in humans to a clinically relevant extent.
Biotransformation
The diastereomers (S,R)-darolutamide and (S,S)-darolutamide are able to interconvert via the metabolite keto-darolutamide with a preference for (S,S)-darolutamide.
Following single oral administration of 300 mg14 C-darolutamide given as an oral solution, keto-darolutamide is the only major metabolite with about 2-fold higher total exposure in plasma compared to darolutamide. Darolutamide and keto-darolutamide accounted together for 87.4% of the 14C-radioactivity in plasma indicating that all other metabolites are of minor importance. Darolutamide is metabolised primarily by oxidative metabolism mediated mainly by CYP3A4, as well as by direct glucuronidation mediated preferentially by UGT1A9 and UGT1A1. In addition, mainly the AKR1C isoforms were shown to catalyse the reduction of keto-darolutamide to the substance diastereomers.
Elimination
The effective half-life of darolutamide and keto-darolutamide in plasma of patients is approximately 18 to 20 hours. Of the two diastereomers comprising darolutamide, (S,R)-darolutamide has a shorter effective half-life of 9 hours compared to (S,S)-darolutamide with an effective half-life of 22 hours. The clearance of darolutamide following intravenous administration was 116 mL/min (CV: 39.7%). A total of 63.4% of substance-related material is excreted in the urine (approximately 7% unchanged), 32.4% is excreted in the faeces. More than 95% of the dose was recovered within 7 days after administration.
Linearity/Non-linearity
In the dose range of 100 to 700 mg (after single dose and at steady state), the exposure to the two diastereomers and the major metabolite keto-darolutamide increases linearly in a nearly dose-related manner. Based on a saturated absorption, no further increase in exposure to darolutamide was observed at 900 mg twice daily.
Special populations
Elderly
No clinically relevant differences in the pharmacokinetics of darolutamide were observed (65-95 years).
Renal impairment
In a clinical pharmacokinetic study, AUC and Cmax for darolutamide were 2.5 and 1.6-fold higher in patients with severe renal impairment (estimated Glomerular Filtration Rate [eGFR] 15 to 29 mL/min/1.73 m²) compared to healthy volunteers.
A population pharmacokinetic analysis indicates a 1.1-, 1.3- and an approximately 1.5-fold higher exposure (AUC) of darolutamide in patients with mild, moderate and severe renal impairment (eGFR 15 to 89 mL/min/1.73 m²) compared to patients with normal renal function.
The pharmacokinetics of darolutamide has not been studied in patients with end-stage renal disease receiving dialysis (eGFR <15 mL/min/1.73 m²).
Hepatic impairment
In a clinical pharmacokinetic study, Cmax and AUC for darolutamide were 1.5 and 1.9-fold higher in patients with moderate hepatic impairment (Child-Pugh B) compared to healthy volunteers. There are no data for patients with severe hepatic impairment (Child-Pugh C).
Ethnic differences
No clinically relevant differences in the pharmacokinetics of darolutamide were observed based on ethnicity (White, Japanese, non-Japanese Asian, Black or African American). A population pharmacokinetic analysis indicated a geometric mean increase in exposure (AUC) of up to 1.56-fold (90% CI: 1.43 to 1.70) in Japanese patients compared to patients from all other regions in both the ARAMIS and ARASENS studies.
5.3. Preclinical safety data
Systemic toxicity
In repeated dose toxicity studies in rats and dogs, the main findings were changes in the male reproductive organs (decreases in organ weight with atrophy of the prostate and epididymides). These effects occurred at systemic exposures in the range of or below the anticipated human exposure (based on AUC comparison). Additional changes to reproductive tissues included minimal increase in vacuolation of the pituitary gland, atrophy and secretory reduction in seminal vesicles and mammary glands in rats as well as testicular hypospermia, seminiferous tubule dilatation and degeneration in dogs. Changes in the male reproductive organs in both species were consistent with the pharmacological activity of darolutamide and reversed or partially resolved after 4- to 8-week recovery periods.
Embryotoxicity/teratogenicity
Studies on developmental toxicity have not been performed.
Reproduction toxicity
Studies on reproductive toxicity have not been performed. However, male fertility is likely to be impaired based on the findings in repeat-dose toxicity studies in rats and dogs, which are consistent with the pharmacological activity of darolutamide.
Genotoxicity and carcinogenicity
Darolutamide did not induce mutations in the microbial mutagenesis (Ames) assay. At high concentrations, darolutamide did induce structural chromosome aberrations in vitro in cultured human lymphocytes. However, in the in vivo combined bone marrow micronucleus test and the Comet assay in the liver and duodenum of the rat, no genotoxicity was observed at exposures in excess of the maximum human exposure.
Oral administration of darolutamide to male rasH2 transgenic mice for 6 months did not show carcinogenic potential at doses up to 1000 mg/kg/day, which is 0.9-1.3 times for darolutamide and 2.1-2.3 times for keto-darolutamide the clinical exposure (AUC) at the recommended clinical daily dose of 1200 mg/day. Based on this study carcinogenic risk of darolutamide cannot be completely excluded.
Safety pharmacology
In vitro, darolutamide weakly inhibited the hERG potassium current and the L-type calcium channel. In vivo, in anaesthetised dogs, darolutamide slightly decreased the QT interval duration, but this effect was not found in conscious dogs.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.