OFEV Soft capsule Ref.[8917] Active ingredients: Nintedanib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Boehringer Ingelheim International GmbH, Binger Strasse 173, 55216, Ingelheim am Rhein, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors
ATC code: L01EX09
Mechanism of action
Nintedanib is a small molecule tyrosine kinase inhibitor including the receptors platelet-derived growth factor receptor (PDGFR) α and β, fibroblast growth factor receptor (FGFR) 1-3, and VEGFR 1-3. In addition, nintedanib inhibits Lck (lymphocyte-specific tyrosine-protein kinase), Lyn (tyrosine-protein kinase lyn), Src (proto-oncogene tyrosine-protein kinase src), and CSF1R (colony stimulating factor 1 receptor) kinases. Nintedanib binds competitively to the adenosine triphosphate (ATP) binding pocket of these kinases and blocks the intracellular signalling cascades, which have been demonstrated to be involved in the pathogenesis of fibrotic tissue remodelling in interstitial lung diseases.
Pharmacodynamic effects
In in vitro studies using human cells nintedanib has been shown to inhibit processes assumed to be involved in the initiation of the fibrotic pathogenesis, the release of pro-fibrotic mediators from peripheral blood monocytic cells and macrophage polarisation to alternatively activated macrophages. Nintedanib has been demonstrated to inhibit fundamental processes in organ fibrosis, proliferation and migration of fibroblasts and transformation to the active myofibroblast phenotype and secretion of extracellular matrix. In animal studies in multiple models of IPF, SSc/SSc-ILD, rheumatoid arthritis-associated-(RA-)ILD and other organ fibrosis, nintedanib has shown anti-inflammatory effects and anti-fibrotic effects in the lung, skin, heart, kidney, and liver. Nintedanib also exerted vascular activity. It reduced dermal microvascular endothelial cell apoptosis and attenuated pulmonary vascular remodelling by reducing the proliferation of vascular smooth muscle cells, the thickness of pulmonary vessel walls and percentage of occluded pulmonary vessels.
Clinical efficacy and safety
Idiopathic pulmonary fibrosis (IPF)
The clinical efficacy of nintedanib has been studied in patients with IPF in two phase III, randomised, double-blind, placebo-controlled studies with identical design (INPULSIS-1 (1 199.32) and INPULSIS-2 (1 199.34)). Patients with FVC baseline <50% predicted or carbon monoxide diffusing capacity (DLCO, corrected for haemoglobin) <30% predicted at baseline were excluded from the trials. Patients were randomized in a 3:2 ratio to treatment with Ofev 150 mg or placebo twice daily for 52 weeks.
The primary endpoint was the annual rate of decline in forced vital capacity (FVC). The key secondary endpoints were change from baseline in Saint George’s Respiratory Questionnaire (SGRQ) total score at 52 weeks and time to first acute IPF exacerbation.
Annual rate of decline in FVC
The annual rate of decline of FVC (in mL) was significantly reduced in patients receiving nintedanib compared to patients receiving placebo. The treatment effect was consistent in both trials. See Table 3 for individual and pooled study results.
Table 3. Annual rate of decline in FVC (mL) in trials INPULSIS-1, INPULSIS-2 and their pooled data – treated set:
| INPULSIS-1 | INPULSIS-2 | INPULSIS-1 and INPULSIS-2 pooled | ||||
|---|---|---|---|---|---|---|
| Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | |
| Number of analysed patients | 204 | 309 | 219 | 329 | 423 | 638 |
| Rate1 (SE) of decline over 52 weeks | -239.9 (18.71) | -114.7 (15.33) | -207.3 (19.31) | -113.6 (15.73) | -223.5 (13.45) | -113.6 (10.98) |
| Comparison vs placebo | ||||||
| Difference1 | 125.3 | 93.7 | 109.9 | |||
| 95% CI | (77.7, 172.8) | (44.8, 142.7) | ||||
| p-value | <0.0001 | 0.0002 | <0.0001 | |||
1 Estimated based on a random coefficient regression model.
CI: confidence interval
In a sensitivity analysis which assumed that in patients with missing data at week 52 the FVC decline after the last observed value would be the same as in all placebo patients, the adjusted difference in the annual rate of decline between nintedanib and placebo was 113.9 mL/year (95% CI 69.2, 158.5) in INPULSIS-1 and 83.3 mL/year (95% CI 37.6, 129.0) in INPULSIS-2.
See Figure 1 for the evolution of change from baseline over time in both treatment groups, based on the pooled analysis of studies INPULSIS-1 and INPULSIS-2.
Figure 1. Mean (SEM) observed FVC change from baseline (mL) over time, studies INPULSIS-1 and INPULSIS-2 pooled:

bid = twice daily
FVC responder analysis
In both INPULSIS trials, the proportion of FVC responders, defined as patients with an absolute decline in FVC % predicted no greater than 5% (a threshold indicative of the increasing risk of mortality in IPF), was significantly higher in the nintedanib group as compared to placebo. Similar results were observed in analyses using a conservative threshold of 10%. See Table 4 for individual and pooled study results.
Table 4. Proportion of FVC responders at 52 weeks in trials INPULSIS-1, INPULSIS-2 and their pooled data – treated set:
| INPULSIS-1 | INPULSIS-2 | INPULSIS-1 and INPULSIS-2 pooled | ||||
|---|---|---|---|---|---|---|
| Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | |
| Number of analysed patients | 204 | 309 | 219 | 329 | 423 | 638 |
| 5% threshold | ||||||
| Number (%) of FVC responders1 | 78 (38.2) | 163 (52.8) | 86 (39.3) | 175 (53.2) | 164 (38.8) | 338 (53.0) |
| Comparison vs placebo | ||||||
| Odds ratio | 1.85 | 1.79 | 1.84 | |||
| 95% CI | (1.28, 2.66) | (1.26, 2.55) | (1.43, 2.36) | |||
| p-value2 | 0.0010 | 0.0011 | <0.0001 | |||
| 10% threshold | ||||||
| Number (%) of FVC responders1 | 116 (56.9) | 218 (70.6) | 140 (63.9) | 229 (69.6) | 256 (60.5) | 447 (70.1) |
| Comparison vs placebo | ||||||
| Odds ratio | 1.91 | 1.29 | 1.58 | |||
| 95% CI | (1.32, 2.79) | (0.89, 1.86) | (1.21, 2.05) | |||
| p-value2 | 0.0007 | 0.1833 | 0.0007 | |||
1 Responder patients are those with no absolute decline greater than 5% or greater than 10% in FVC % predicted, depending on the threshold and with an FVC evaluation at 52 weeks.
2 Based on a logistic regression.
Time to progression (≥10% absolute decline of FVC % predicted or death)
In both INPULSIS trials, the risk of progression was statistically significantly reduced for patients treated with nintedanib compared with placebo. In the pooled analysis, the HR was 0.60 indicating a 40% reduction in the risk of progression for patients treated with nintedanib compared with placebo.
Table 5. Frequency of patients with ≥10% absolute decline of FVC % predicted or death over 52 weeks and time to progression in trials INPULSIS-1, INPULSIS-2, and their pooled data – treated set:
| INPULSIS-1 | INPULSIS-2 | INPULSIS-1 and INPULSIS-2 pooled | ||||
|---|---|---|---|---|---|---|
| Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | |
| Number at risk | 204 | 309 | 219 | 329 | 423 | 638 |
| Patients with events, N (%) | 83 (40.7) | 75 (24.3) | 92 (42.0) | 98 (29.8) | 175 (41.4) | 173 (27.1) |
| Comparison vs placebo1 | ||||||
| p-value2 | 0.0001 | 0.0054 | <0.0001 | |||
| Hazard ratio3 | 0.53 | 0.67 | 0.60 | |||
| 95% CI | (0.39, 0.72) | (0.51, 0.89) | (0.49, 0.74) | |||
1 Based on data collected up to 372 days (52 weeks + 7 day margin).
2 Based on a Log-rank test.
3 Based on a Cox’s regression model.
Change from baseline in SGRQ total score at week 52
In the pooled analysis of the INPULSIS trials, the baseline SGRQ scores were 39.51 in the nintedanib group and 39.58 in the placebo group. The estimated mean change from baseline to week 52 in SGRQ total score was smaller in the nintedanib group (3.53) than in the placebo group (4.96), with a difference between the treatment groups of -1.43 (95% CI: -3.09, 0.23; p=0.0923). Overall, the effect of nintedanib on health-related quality of life as measured by the SGRQ total score is modest, indicating less worsening compared to placebo.
Time to first acute IPF exacerbation
In the pooled analysis of the INPULSIS trials, a numerically lower risk of first acute exacerbation was observed in patients receiving nintedanib compared to placebo. See Table 6 for individual and pooled study results.
Table 6. Frequency of patients with acute IPF exacerbations over 52 weeks and time to first exacerbation analysis based on investigator-reported events in trials INPULSIS-1, INPULSIS-2, and their pooled data – treated set:
| INPULSIS-1 | INPULSIS-2 | INPULSIS-1 and INPULSIS-2 pooled | ||||
|---|---|---|---|---|---|---|
| Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | |
| Number at risk | 204 | 309 | 219 | 329 | 423 | 638 |
| Patients with events, N (%) | 11 (5.4) | 19 (6.1) | 21 (9.6) | 12 (3.6) | 32 (7.6) | 31 (4.9) |
| Comparison vs placebo1 | ||||||
| p-value2 | 0.6728 | 0.0050 | 0.0823 | |||
| Hazard ratio3 | 1.15 | 0.38 | 0.64 | |||
| 95% CI | (0.54, 2.42) | (0.19, 0.77) | (0.39, 1.05) | |||
1 Based on data collected up to 372 days (52 weeks + 7 day margin).
2 Based on a Log-rank test.
3 Based on a Cox’s regression model.
In a pre-specified sensitivity analysis, the frequency of patients with at least 1 adjudicated exacerbation occurring within 52 weeks was lower in the nintedanib group (1.9% of patients) than in the placebo group (5.7% of patients). Time to event analysis of the adjudicated exacerbation events using pooled data yielded a hazard ratio (HR) of 0.32 (95% CI 0.16, 0.65; p=0.0010).
Survival analysis
In the pre-specified pooled analysis of survival data of the INPULSIS trials, overall mortality over 52 weeks was lower in the nintedanib group (5.5%) compared with the placebo group (7.8%). The analysis of time to death resulted in a HR of 0.70 (95% CI 0.43, 1.12; p=0.1399). The results of all survival endpoints (such as on-treatment mortality and respiratory mortality) showed a consistent numerical difference in favour of nintedanib.
Table 7. All-cause mortality over 52 weeks in trials INPULSIS-1, INPULSIS-2, and their pooled data – treated set:
| INPULSIS-1 | INPULSIS-2 | INPULSIS-1 and INPULSIS-2 pooled | ||||
|---|---|---|---|---|---|---|
| Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | Placebo | Ofev 150 mg twice daily | |
| Number at risk | 204 | 309 | 219 | 329 | 423 | 638 |
| Patients with events, N (%) | 13 (6.4) | 13 (4.2) | 20 (9.1) | 22 (6.7) | 33 (7.8) | 35 (5.5) |
| Comparison vs placebo1 | ||||||
| p-value2 | 0.2880 | 0.2995 | 0.1399 | |||
| Hazard ratio3 | 0.63 | 0.74 | 0.70 | |||
| 95% CI | (0.29, 1.36) | (0.40, 1.35) | (0.43, 1.12) | |||
1 Based on data collected up to 372 days (52 weeks + 7 day margin).
2 Based on a Log-rank test.
3 Based on a Cox’s regression model.
Long-term treatment with Ofev in patients with IPF (INPULSIS-ON)
An open-label extension trial of Ofev included 734 patients with IPF. Patients who completed the 52-week treatment period in an INPULSIS trial received open-label Ofev treatment in the extension trial INPULSIS-ON. Median exposure time for patients treated with Ofev in both the INPULSIS and INPULSIS-ON trials was 44.7 months (range 11.9-68.3). The exploratory efficacy endpoints included the annual rate of decline in FVC over 192 weeks which was -135.1 (5.8) mL/year in all patients treated and were consistent with the annual rate of FVC decline in patients treated with Ofev in the INPULSIS phase III trials (-113.6 mL per year). The adverse event profile of Ofev in INPULSIS-ON was consistent to that in the INPULSIS phase III trials.
IPF patients with advanced lung function impairment (INSTAGE)
INSTAGE was a multicentre, multinational, prospective, randomised, double-blind, parallel-group clinical trial in IPF patients with advanced lung function impairment (DLCO ≤35% predicted) for 24 weeks. 136 patients were treated with Ofev monotherapy. Primary endpoint result showed a reduction of St Georges Respiratory Questionnaire (SGRQ) total score by -0.77 units at week W12, based on adjusted mean change from baseline. A post hoc comparison demonstrated that the decline in FVC in these patients was consistent with the decline in FVC in patients with less advanced disease and treated with Ofev in the INPULSIS phase III trials.
The safety and tolerability profile of Ofev in IPF patients with advanced lung function impairment was consistent with that seen in the INPULSIS phase III trials.
Additional data from the phase IV INJOURNEY trial with Ofev 150 mg twice daily and add-on pirfenidone
Concomitant treatment with nintedanib and pirfenidone has been investigated in an exploratory open-label, randomised trial of nintedanib 150 mg twice daily with add-on pirfenidone (titrated to 801 mg three times a day) compared to nintedanib 150 mg twice daily alone in 105 randomised patients for 12 weeks. The primary endpoint was the percentage of patients with gastrointestinal adverse events from baseline to week 12. Gastrointestinal adverse events were frequent and in line with the established safety profile of each component. Diarrhoea, nausea and vomiting were the most frequent adverse events reported in patients, treated with pirfenidone added to nintedanib versus nintedanib alone, respectively.
Mean (SE) absolute changes from baseline in FVC at week 12 were -13.3 (17.4) mL in patients treated with nintedanib with add-on pirfenidone (n=48) compared to -40.9 (31.4) mL in patients treated with nintedanib alone (n=44).
Other chronic fibrosing interstitial lung diseases (ILDs) with a progressive phenotype
The clinical efficacy of Ofev has been studied in patients with other chronic fibrosing ILDs with a progressive phenotype in a double-blind, randomised, placebo-controlled phase III trial (INBUILD). Patients with IPF were excluded. Patients with a clinical diagnosis of a chronic fibrosing ILD were selected if they had relevant fibrosis (greater than 10% fibrotic features) on HRCT and presented with clinical signs of progression (defined as FVC decline ≥10%, FVC decline ≥5% and <10% with worsening symptoms or imaging, or worsening symptoms and worsening imaging all in the 24 months prior to screening). Patients were required to have an FVC greater than or equal to 45% of predicted and a DLCO 30% to less than 80% of predicted. Patients were required to have progressed despite management deemed appropriate in clinical practice for the patient’s relevant ILD.
A total of 663 patients were randomised in a 1:1 ratio to receive either Ofev 150 mg bid or matching placebo for at least 52 weeks. The median Ofev exposure over the whole trial was 17.4 months and the mean Ofev exposure over the whole trial was 15.6 months. Randomisation was stratified based on HRCT fibrotic pattern as assessed by central readers. 412 patients with HRCT with usual interstitial pneumonia (UIP)-like fibrotic pattern and 251 patients with other HRCT fibrotic patterns were randomised. There were 2 co-primary populations defined for the analyses in this trial: all patients (the overall population) and patients with HRCT with UIP-like fibrotic pattern. Patients with other HRCT fibrotic patterns represented the ‘complementary’ population.
The primary endpoint was the annual rate of decline in forced vital capacity (FVC) (in mL) over 52 weeks. Main secondary endpoints were absolute change from baseline in King’s Brief Interstitial Lung Disease Questionnaire (K-BILD) total score at week 52, time to first acute ILD exacerbation or death over 52 weeks, and time to death over 52 weeks.
Patients had a mean (standard deviation [SD, Min-Max]) age of 65.8 (9.8, 27-87) years and a mean FVC percent predicted of 69.0% (15.6, 42-137). The underlying clinical ILD diagnoses in groups represented in the trial were hypersensitivity pneumonitis (26.1%), autoimmune ILDs (25.6%), idiopathic nonspecific interstitial pneumonia (18.9%), unclassifiable idiopathic interstitial pneumonia (17.2%), and other ILDs (12.2%).
The INBUILD trial was not designed or powered to provide evidence for a benefit of nintedanib in specific diagnostic subgroups. Consistent effects were demonstrated in subgroups based on the ILD diagnoses. The experience with nintedanib in very rare progressive fibrosing ILDs is limited.
Annual rate of decline in FVC
The annual rate of decline in FVC (in mL) over 52 weeks was significantly reduced by 107.0 mL in patients receiving Ofev compared to patients receiving placebo (Table 8) corresponding to a relative treatment effect of 57.0%.
Table 8. Annual rate of decline in FVC (mL) over 52 weeks:
| Placebo | Ofev 150 mg twice daily | |
|---|---|---|
| Number of analysed patients | 331 | 332 |
| Rate1 (SE) of decline over 52 weeks | -187.8 (14.8) | -80.8 (15.1) |
| Comparison vs placebo | ||
| Difference1 | 107.0 | |
| 95% CI | (65.4, 148.5) | |
| p-value | <0.0001 | |
1 Based on a random coefficient regression with fixed categorical effects of treatment, HRCT pattern, fixed continuous effects of time, baseline FVC [mL], and including treatment-by-time and baseline-by-time interactions
Similar results were observed in the co-primary population of patients with HRCT with UIP-like fibrotic pattern. The treatment effect was consistent in the complementary population of patients with other HRCT fibrotic patterns (interaction p-value 0.2268) (Figure 2).
Figure 2. Forest plot of the annual rate of decline in FVC (mL) over 52 weeks in the patient populations:
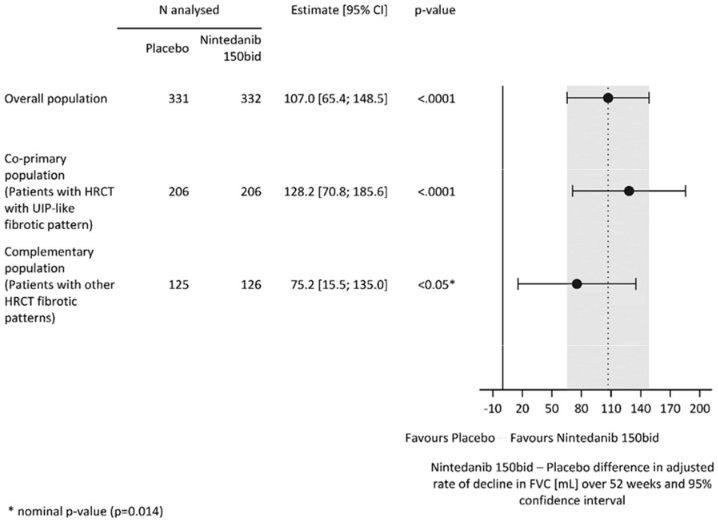
bid = twice daily
The results of the effect of Ofev in reducing the annual rate of decline in FVC were confirmed by all pre-specified sensitivity analyses and consistent results were observed in the pre-specified efficacy subgroups: gender, age group, race, predicted baseline FVC %, and original underlying clinical ILD diagnosis in groups.
Figure 3 shows the evolution of change in FVC from baseline over time in the treatment groups.
Figure 3. Mean (SEM) observed FVC change from baseline (mL) over 52 weeks:
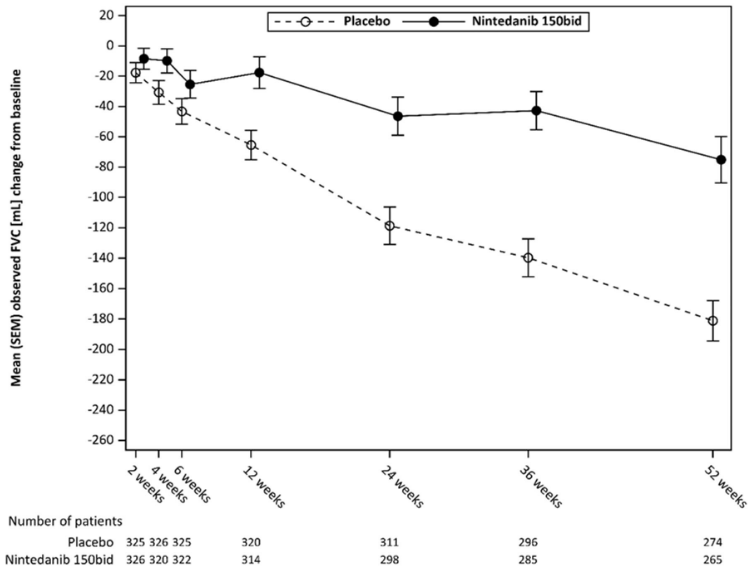
bid = twice daily
In addition, favourable effects of Ofev were observed on the adjusted mean absolute change from baseline in FVC % predicted at week 52. The adjusted mean absolute change from baseline to week 52 in FVC % predicted was lower in the nintedanib group (-2.62%) than in the placebo group (-5.86%). The adjusted mean difference between the treatment groups was 3.24 (95% CI: 2.09, 4.40, nominal p<0.0001).
FVC responder analysis
The proportion of FVC responders, defined as patients with a relative decline in FVC % predicted no greater than 5%, was higher in the Ofev group as compared to placebo. Similar results were observed in analyses using a threshold of 10% (Table 9).
Table 9. Proportion of FVC responders at 52 weeks in INBUILD:
| Placebo | Ofev 150 mg twice daily | |
|---|---|---|
| Number of analysed patients | 331 | 332 |
| 5% threshold | ||
| Number (%) of FVC responders1 | 104 (31.4) | 158 (47.6) |
| Comparison vs placebo | ||
| Odds ratio2 | 2.01 | |
| 95% CI | (1.46, 2.76) | |
| Nominal p-value | <0.0001 | |
| 10% threshold | ||
| Number (%) of FVC responders1 | 169 (51.1) | 197 (59.3) |
| Comparison vs placebo | ||
| Odds ratio2 | 1.42 | |
| 95% CI | (1.04, 1.94) | |
| Nominal p-value | 0.0268 | |
1 Responder patients are those with no relative decline greater than 5% or greater than 10% in FVC % predicted, depending on the threshold and with an FVC evaluation at 52 weeks (patients with missing data at week 52 were considered as non-responders).
2 Based on a logistic regression model with continuous covariate baseline FVC % predicted and binary covariate HRCT pattern.
Time to first acute ILD exacerbation or death
Over the whole trial, the proportion of patients with at least one event of first acute ILD exacerbation or death was 13.9% in the Ofev group and 19.6% in the placebo group. The HR was 0.67 (95% CI: 0.46, 0.98; nominal p=0.0387), indicating a 33% reduction in the risk of first acute ILD exacerbation or death in patients receiving Ofev compared to placebo (Figure 4).
Figure 4. Kaplan-Meier plot of time to first acute ILD exacerbation or death over the whole trial:
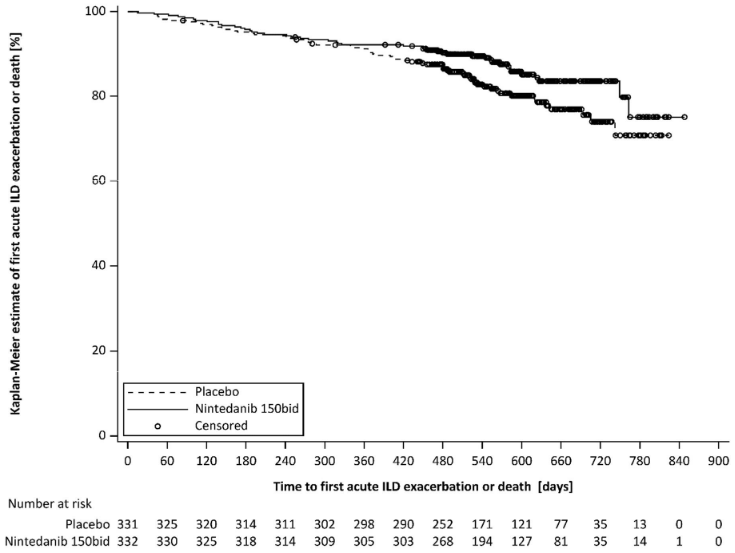
bid = twice daily
Survival analysis
The risk of death was lower in the Ofev group compared to the placebo group. The HR was 0.78 (95% CI: 0.50, 1.21; nominal p=0.2594), indicating a 22% reduction in the risk of death in patients receiving Ofev compared to placebo.
Time to progression (≥10% absolute decline of FVC % predicted) or death
In the INBUILD trial, the risk of progression (≥10% absolute decline of FVC % predicted) or death was reduced for patients treated with Ofev. The proportion of patients with an event was 40.4% in the Ofev group and 54.7% in the placebo group. The HR was 0.66 (95% CI: 0.53, 0.83; p=0.0003), indicating a 34% reduction of the risk of progression (≥10% absolute decline of FVC % predicted) or death in patients receiving Ofev compared to placebo.
Quality of life
The adjusted mean change from baseline in K-BILD total score at week 52 was -0.79 units in the placebo group and 0.55 in the Ofev group. The difference between the treatment groups was 1.34 (95% CI: -0.31, 2.98; nominal p=0.1115).
The adjusted mean absolute change from baseline in Living with Pulmonary Fibrosis (L-PF) symptoms dyspnoea domain score at week 52 was 4.28 in the Ofev group compared with 7.81 in the placebo group. The adjusted mean difference between the groups in favour of Ofev was -3.53 (95% CI: -6.14, -0.92; nominal p=0.0081). The adjusted mean absolute change from baseline in L-PF Symptoms cough domain score at week 52 was -1.84 in the Ofev group compared with 4.25 in the placebo group. The adjusted mean difference between the groups in favour of Ofev was -6.09 (95% CI: -9.65, -2.53; nominal p=0.0008).
Systemic sclerosis associated interstitial lung disease (SSc-ILD)
The clinical efficacy of Ofev has been studied in patients with SSc-ILD in a double-blind, randomised, placebo-controlled phase III trial (SENSCIS). Patients were diagnosed with SSc-ILD based upon the 2013 American College of Rheumatology/European League Against Rheumatism classification criteria for SSc and a chest high resolution computed tomography (HRCT) scan conducted within the previous 12 months. A total of 580 patients were randomised in a 1:1 ratio to receive either Ofev 150 mg bid or matching placebo for at least 52 weeks, of which 576 patients were treated. Randomisation was stratified by antitopoisomerase antibody status (ATA). Individual patients stayed on blinded trial treatment for up to 100 weeks (median Ofev exposure 15.4 months; mean Ofev exposure 14.5 months).
The primary endpoint was the annual rate of decline in FVC over 52 weeks. Key secondary endpoints were absolute change from baseline in the modified Rodnan Skin Score (mRSS) at week 52 and absolute change from baseline in the Saint George’s Respiratory Questionnaire (SGRQ) total score at week 52.
In the overall population, 75.2% of the patients were female. The mean (standard deviation [SD, Min-Max]) age was 54.0 (12.2, 20-79) years. Overall, 51.9% of patients had diffuse cutaneous systemic sclerosis (SSc) and 48.1% had limited cutaneous SSc. The mean (SD) time since first onset of a non- Raynaud symptom was 3.49 (1.7) years. 49.0% of patients were on stable therapy with mycophenolate at baseline (46.5% mycophenolate mofetil, 1.9% mycophenolate sodium, 0.5% mycophenolic acid). The safety profile in patients with or without mycophenolate at baseline was comparable.
Annual rate of decline in FVC
The annual rate of decline of FVC (mL) over 52 weeks was significantly reduced by 41.0 mL in patients receiving Ofev compared to patients receiving placebo (Table 10) corresponding to a relative treatment effect of 43.8%.
Table 10. Annual rate of decline in FVC (mL) over 52 weeks:
| Placebo | Ofev 150 mg twice daily | |
|---|---|---|
| Number of analysed patients | 288 | 287 |
| Rate1 (SE) of decline over 52 weeks | -93.3 (13.5) | -52.4 (13.8) |
| Comparison vs placebo | ||
| Difference1 | 41.0 | |
| 95% CI | (2.9, 79.0) | |
| p-value | <0.05 | |
1 Based on a random coefficient regression with fixed categorical effects of treatment, ATA status, gender, fixed continuous effects of time, baseline FVC [mL], age, height, and including treatment-by-time and baseline-by-time interactions. Random effect was included for patient specific intercept and time. Within-patient errors were modelled by an unstructured variance-covariance matrix. Inter-individual variability was modelled by a variance-components variance-covariance matrix.
The effect of Ofev in reducing the annual rate of decline in FVC was similar across pre-specified sensitivity analyses and no heterogeneity was detected in pre-specified subgroups (e.g. by age, gender, and mycophenolate use).
In addition, similar effects were observed on other lung function endpoints, e.g absolute change from baseline in FVC in mL at week 52 (Figure 5 and Table 11) and rate of decline in FVC in % predicted over 52 weeks (Table 12) providing further substantiation of the effects of Ofev on slowing progression of SSc-ILD. Furthermore, fewer patients in the Ofev group had an absolute FVC decline >5% predicted (20.6% in the Ofev group vs. 28.5% in the placebo group, OR=0.65, p=0.0287). The relative FVC decline in mL >10% was comparable between both groups (16.7% in the Ofev group vs. 18.1% in the placebo group, OR=0.91, p=0.6842). In these analyses, missing FVC values at week 52 were imputed with the patient’s worst value on treatment.
An exploratory analysis of data up to 100 weeks (maximum treatment duration in SENSCIS) suggested that the on treatment effect of Ofev on slowing progression of SSc-ILD persisted beyond 52 weeks.
Figure 5. Mean (SEM) observed FVC change from baseline (mL) over 52 weeks:
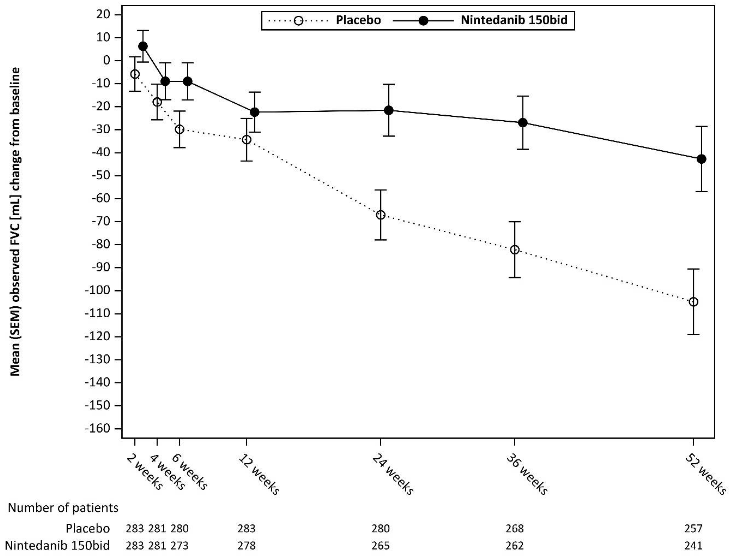
bid = twice daily
Table 11. Absolute change from baseline in FVC (mL) at week 52:
| Placebo | Ofev 150 mg twice daily | |
|---|---|---|
| Number of analysed patients | 288 | 288 |
| Mean (SD) at Baseline | 2 541.0 (815.5) | 2 458.5 (735.9) |
| Mean1 (SE) change from baseline at week 52 | -101.0 (13.6) | -54.6 (13.9) |
| Comparison vs placebo | ||
| Mean1 | 46.4 | |
| 95% CI | (8.1, 84.7) | |
| p-value | <0.05 | |
1 Based on Mixed Model for Repeated Measures (MMRM), with fixed categorical effects of ATA status, visit, treatment-by-visit interaction, baseline-by-visit interaction age, gender and height. Visit was the repeated measure. Within-patient errors were modelled by unstructured variance-covariance structure. Adjusted mean was based on all analysed patients in the model (not only patients with a baseline and measurement at week 52).
Table 12. Annual rate of decline in FVC (% predicted) over 52 weeks:
| Placebo | Ofev 150 mg twice daily | |
|---|---|---|
| Number of analysed patients | 288 | 287 |
| Rate1 (SE) of decline over 52 weeks | -2.6 (0.4) | -1.4 (0.4) |
| Comparison vs placebo | ||
| Difference1 | 1.15 | |
| 95% CI | (0.09, 2.21) | |
| p-value | <0.05 | |
1 Based on a random coefficient regression with fixed categorical effects of treatment, ATA status, fixed continuous effects of time, baseline FVC [% pred], and including treatment-by-time and baseline-by-time interactions. Random effect was included for patient specific intercept and time. Within-patient errors were modelled by an unstructured variance-covariance matrix. Inter-individual variability was modelled by a variance-components variance-covariance matrix
Change from baseline in Modified Rodnan Skin Score (mRSS) at week 52
The adjusted mean absolute change from baseline in mRSS at week 52 was comparable between the Ofev group (-2.17 (95% CI -2.69, -1.65)) and the placebo group (-1.96 (95% CI -2.48, -1.45)). The adjusted mean difference between the treatment groups was -0.21 (95% CI -0.94, 0.53; p=0.5785).
Change from baseline in St. George’s Respiratory Questionnaire (SGRQ) total score at week 52 The adjusted mean absolute change from baseline in SGRQ total score at week 52 was comparable between the Ofev group (0.81 (95% CI -0.92, 2.55)) and the placebo group (-0.88 (95% CI -2.58, 0.82)). The adjusted mean difference between the treatment groups was 1.69 (95% CI -0.73, 4.12; p=0.1711).
Survival analysis
Mortality over the whole trial was comparable between the Ofev group (N=10; 3.5%) and the placebo group (N=9; 3.1%). The analysis of time to death over the whole trial resulted in a HR of 1.16 (95% CI 0.47, 2.84; p = 0.7535).
QT interval
In a dedicated study in renal cell cancer patients, QT/QTc measurements were recorded and showed that a single oral dose of 200 mg nintedanib as well as multiple oral doses of 200 mg nintedanib administered twice daily for 15 days did not prolong the QTcF interval.
Paediatric population
Fibrosing interstitial lung diseases (ILDs) in children and adolescents
The clinical safety and efficacy of Ofev in children and adolescents from 6 to 17 years with clinically significant fibrosing interstitial lung diseases (ILDs) was assessed in an exploratory randomised, double-blind, placebo-controlled phase III trial (InPedILD 1199.337) (see section 4.2).
The InPedILD trial enrolled children and adolescents aged 6 to 17 years with clinically significant fibrosing ILD and FVC of at least 25% predicted. Patients were classified as having fibrosing ILD based on evidence of fibrosis on two HRCT scans (with one HRCT scan conducted within the previous 12 months) or evidence of fibrosis on lung biopsy and one HRCT scan conducted within the previous 12 months.
Clinically significant disease was defined as a Fan score ≥3 or documented evidence of clinical progression over any time frame. Evidence of clinical progression was based on a relative decline in FVC ≥10% predicted, a relative decline in FVC of 5–10% predicted with worsening symptoms, worsening fibrosis on HRCT or other measures of clinical worsening attributed to progressive pulmonary fibrosis (e.g. increased oxygen requirement, decreased diffusion capacity) although this was not a requirement for enrolment for patients with a Fan score of ≥3.
Patients were randomised in a 2:1 ratio to receive either Ofev twice daily (doses adjusted for weight, including the use of a 25 mg capsule) or matching placebo for 24 weeks, followed by open label treatment with nintedanib of variable duration.The use of standard of care as deemed clinically indicated by the treating physician was allowed.
In total, 39 patients were randomised (61.5% female), (6-11 years: 12 patients, 12-17 years: 27 patients). The mean [standard deviation (SD)] age was 12.6 (3.3) years. Mean (SD) weight was 42.2 kg (17.8 kg); 6-11 years: 26.6 kg (10.4 kg), 12-17 years: 49.1 kg (16.0 kg). Trial 1199-0337 enrolled patients with a broad spectrum of diseases. The most frequent single underlying ILD diagnoses were ‘surfactant protein deficiency’ (nintedanib: 26.9%, placebo: 38.5%), ‘systemic sclerosis’ (nintedanib: 15.4%, placebo: 23.1%), and ‘toxic/radiation/drug-induced pneumonitis’ (nintedanib: 11.5%, placebo 7.7%). Chronic hypersensitivity pneumonitis was reported for 2 patients (nintedanib: 7.7%). The remaining underlying ILD diagnoses reported for 1 patient each were post-HSCT fibrosis, juvenile RA, juvenile idiopathic arthritis, Dermatomyositis (DM), Desquammative Interstitial Pneumonitis, Influenza H1N1, Unclear (Chronic Diffuse Pulmonary Lung Disease), Copa Syndrome, Copa Gene Mutation, Undifferentiated Connective Tissue Disease, Post-Infectious Bronchiolitis Obliterans, Unspecified ILD, Idiopathic and Sting-associated Vasculopathy.
All patients were reported with at least 1 concomitant therapy during the double-blind period. Use of concomitant therapies (baseline, on-treatment, and post-study drug discontinuation therapies) to treat the underlying disease including corticosteroids and immunomodulators was permitted.
The primary endpoints results were:
- The exposure to nintedanib described as AUCτ,ss based on sampling at steady state was broadly similar in children and adolescents and comparable to the AUCτ,ss observed in adults (see section 5.2).
- The percentage of patients with treatment-emergent adverse events at week 24 was 84.6% in the nintedanib group (6-11 years: 75.0%, 12-17 years: 88.9%) and 84.6% in the placebo group (6-11 years: 100%, 12-17 years: 77.8%).
There was no primary efficacy endpoint in the study.
Secondary endpoint for lung function was the change in Forced Vital Capacity (FVC) % predicted from baseline at week 24 and week 52. The adjusted mean change from baseline at week 24 in FVC % predicted was 0.31 (95% CI -2.36, 2.98) in the nintedanib group, and -0.89 (95% CI -4.61, 2.82) in the placebo group, with an adjusted mean (95% CI) difference in FVC % predicted of 1.21 (95% CI -3.40, 5.81) in favour of nintedanib. At week 52, the adjusted mean of the difference in change from baseline in FVC % predicted between treatment groups was 1.77 (95% CI -4.70, 8.25).
For the FVC % predicted endpoint and a number of other exploratory efficacy endpoints, high variability in response to treatment with nintedanib was observed amongst paediatric patients.
Safety secondary endpoints included:
- Percentage of patients with treatment-emergent pathological findings of epiphyseal growth plate, which was similar across the treatment groups at week 24 (7.7% in both treatment groups). Up to week 52, the percentage of patients with pathological findings was nintedanib/nintedanib: 11.5% and placebo/nintedanib: 15.4%.
- Percentage of patients with treatment-emergent pathological findings on dental examination or imaging, which was 46.2% in the nintedanib group and 38.5% in the placebo group up to week 24. Up to week 52, the percentage of patients with pathological findings was nintedanib/nintedanib: 50.0% and placebo/nintedanib: 46.2%.
The European Medicines Agency has waived the obligation to submit the results of studies with Ofev in all subsets of the paediatric population in IPF (see section 4.2 for information on paediatric use). The European Medicines Agency has waived the obligation to submit the results of studies with Ofev in paediatric population below 6 years of age in fibrosing ILDs (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Nintedanib reached maximum plasma concentrations approximately 2-4 h after oral administration as soft gelatine capsule under fed conditions (range 0.5-8 h). The absolute bioavailability of a 100 mg dose was 4.69% (90% CI: 3.615-6.078) in healthy volunteers. Absorption and bioavailability are decreased by transporter effects and substantial first-pass metabolism. Nintedanib exposure increased dose-proportionally in the dose range of 50-450 mg once daily and 150-300 mg twice daily. Steady state plasma concentrations were achieved within one week of dosing at the latest.
After food intake, nintedanib exposure increased by approximately 20% compared to administration under fasted conditions (CI: 95.3-152.5%) and absorption was delayed (median tmax fasted: 2.00 h; fed: 3.98 h).
In an in vitro study, mixing nintedanib capsules with a small amount of apple sauce or chocolate pudding for up to 15 minutes did not have any impact on the pharmaceutical quality. Swelling and deformation of the capsules due to the water uptake of the gelatin capsule shell was observed with longer exposure time to the soft food. Therefore, taking the capsules with soft food would not be expected to alter the clinical effect when taken immediately.
Distribution
Nintedanib follows at least bi-phasic disposition kinetics. After intravenous infusion, a high volume of distribution (Vss: 1 050 L, 45.0% gCV) was observed.
The in vitro protein binding of nintedanib in human plasma was high, with a bound fraction of 97.8%. Serum albumin is considered to be the major binding protein. Nintedanib is preferentially distributed in plasma with a blood to plasma ratio of 0.869.
Biotransformation
The prevalent metabolic reaction for nintedanib is hydrolytic cleavage by esterases resulting in the free acid moiety BIBF 1202. BIBF 1202 is subsequently glucuronidated by uridine 5'-diphospho-glucuronosyltransferase enzymes (UGT) enzymes, namely UGT 1A1, UGT 1A7, UGT 1A8, and UGT 1A10 to BIBF 1202 glucuronide.
Only a minor extent of the biotransformation of nintedanib consisted of CYP pathways, with CYP 3A4 being the predominant enzyme involved. The major CYP-dependent metabolite could not be detected in plasma in the human ADME study. In vitro, CYP-dependent metabolism accounted for about 5% compared to about 25% ester cleavage. Nintedanib, BIBF 1202, and BIBF 1202 glucuronide did not inhibit or induce CYP enzymes in preclinical studies, either. Drug-drug interactions between nintedanib and CYP substrates, CYP inhibitors, or CYP inducers are therefore not expected.
Elimination
Total plasma clearance after intravenous infusion was high (CL: 1 390 mL/min, 28.8% gCV). Urinary excretion of the unchanged active substance within 48 h was about 0.05% of the dose (31.5% gCV) after oral and about 1.4% of the dose (24.2% gCV) after intravenous administration; the renal clearance was 20 mL/min (32.6% gCV). The major route of elimination of drug related radioactivity after oral administration of [14C] nintedanib was via faecal/biliary excretion (93.4% of dose, 2.61% gCV). The contribution of renal excretion to the total clearance was low (0.649% of dose, 26.3% gCV). The overall recovery was considered complete (above 90%) within 4 days after dosing. The terminal half-life of nintedanib was between 10 and 15 h (gCV % approximately 50%).
Linearity/non-linearity
The pharmacokinetics (PK) of nintedanib can be considered linear with respect to time (i.e. single-dose data can be extrapolated to multiple-dose data). Accumulation upon multiple administrations was 1.04-fold for Cmax and 1.38-fold for AUCτ. Nintedanib trough concentrations remained stable for more than one year.
Transport
Nintedanib is a substrate of P-gp. For the interaction potential of nintedanib with this transporter, see section 4.5. Nintedanib was shown to be not a substrate or inhibitor of OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, or MRP-2 in vitro. Nintedanib was also not a substrate of BCRP. Only a weak inhibitory potential on OCT-1, BCRP, and P-gp was observed in vitro which is considered to be of low clinical relevance. The same applies for nintedanib being a substrate of OCT-1.
Population pharmocokinetic analysis in special populations
The PK properties of nintedanib were similar in healthy volunteers, patients with IPF, patients with other chronic fibrosing ILDs with a progressive phenotype, patients with SSc-ILD, and cancer patients. Based on results of a population PK (PopPK) analysis in patients with IPF and non small cell lung cancer (NSCLC) (N=1 191) and descriptive investigations, exposure to nintedanib was not influenced by sex (body weight corrected), mild and moderate renal impairment (estimated by creatinine clearance), alcohol consumption, or P-gp genotype. PopPK analyses indicated moderate effects on exposure to nintedanib depending on age, body weight, and race (see below). Based on the high inter-individual variability of exposure observed moderate effects are considered not clinically relevant (see section 4.4).
Age
Exposure to nintedanib increased linearly with age. AUCτ,ss decreased by 16% for a 45-year old patient and increased by 13% for a 76-year old patient relative to a patient with the median age of 62 years. The age range covered by the analysis was 29 to 85 years; approximately 5% of the population were older than 75 years. Based on a PopPK model, an increase in nintedanib exposure of approximately 20-25% was observed in patients ≥75 years compared with patients under 65 years.
Paediatric population
Based on the analysis of pharmacokinetic data of study InPedILD (1199.337), oral administration of nintedanib according to the weight-based dosing algorithm resulted in exposure within the range observed in adult patients. The observed geometric mean AUCτ,ss (geometric coefficient of variation) exposures were 175 ng/mL·hr (85.1%) and 167 ng/mL·hr (83.6%) in 10 patients aged 6 to 11 years old and 23 patients aged 12 to 17 years old, respectively.
Body weight
An inverse correlation between body weight and exposure to nintedanib was observed. AUCτ,ss increased by 25% for a 50 kg patient (5th percentile) and decreased by 19% for a 100 kg patient (95th percentile) relative to a patient with the median weight of 71.5 kg.
Race
The population mean exposure to nintedanib was 33-50% higher in Chinese, Taiwanese, and Indian patients and 16% higher in Japanese patients while it was 16-22% lower in Koreans compared to Caucasians (body weight corrected). Data from Black individuals were very limited but in the same range as for Caucasians.
Hepatic impairment
In a dedicated single dose phase I study and compared to healthy subjects, exposure to nintedanib based on Cmax and AUC was 2.2-fold higher in volunteers with mild hepatic impairment (Child Pugh A; 90% CI 1.3-3.7 for Cmax and 1.2-3.8 for AUC, respectively). In volunteers with moderate hepatic impairment (Child Pugh B), exposure was 7.6-fold higher based on Cmax (90% CI 4.4-13.2) and 8.7-fold higher (90% CI 5.7-13.1) based on AUC, respectively, compared to healthy volunteers. Subjects with severe hepatic impairment (Child Pugh C) have not been studied.
Concomitant treatment with pirfenidone
In a dedicated pharmacokinetic study, concomitant treatment of nintedanib with pirfenidone was investigated in patients with IPF. Group 1 received a single dose of 150 mg nintedanib before and after uptitration to 801 mg pirfenidone three times a day at steady state (N=20 patients treated). Group 2 received steady state treatment of 801 mg pirfenidone three times a day and had a PK profiling before and after at least 7 days of co-treatment with 150 mg nintedanib twice daily (N=17 patients treated).
In group 1, the adjusted geometric mean ratios (90% confidence interval (CI)) were 93% (57% – 151%) and 96% (70% – 131%) for Cmax and AUC0-tz of nintedanib, respectively (n=12 for intraindividual comparison). In group 2, the adjusted geometric mean ratios (90% CI)) were 97% (86% – 110%) and 95% (86% – 106%) for Cmax,ss and AUCτ,ss of pirfenidone, respectively (n=12 for intraindividual comparison). Based on these results, there is no evidence of a relevant pharmacokinetic drug-drug interaction between nintedanib and pirfenidone when administered in combination (see section 4.4).
Concomitant treatment with bosentan
In a dedicated pharmacokinetic study, concomitant treatment of Ofev with bosentan was investigated in healthy volunteers. Subjects received a single dose of 150 mg Ofev before and after multiple dosing of 125 mg bosentan twice daily at steady state. The adjusted geometric mean ratios (90% confidence interval (CI)) were 103% (86% – 124%) and 99% (91% – 107%) for Cmax and AUC0-tz of nintedanib, respectively (n=13), indicating that co-administration of nintedanib with bosentan did not alter the pharmacokinetics of nintedanib.
Concomitant treatment with oral hormonal contraceptives
In a dedicated pharmacokinetic study, female patients with SSc-ILD received a single dose of a combination of 30 μg ethinylestradiol and 150 μg levonorgestrel before and after twice daily dosing of 150 mg nintedanib for at least 10 days. The adjusted geometric mean ratios (90% confidence interval (CI)) were 117% (108% – 127%; Cmax) and 101% (93% – 111%; AUC0-tz) for ethinylestradiol and 101% (90% – 113%; Cmax) and 96% (91% – 102%; AUC0-tz) for levonorgestrel, respectively (n=15), indicating that co-administration of nintedanib has no relevant effect on the plasma exposure of ethinylestradiol and levonorgestrel.
Exposure-response relationship
Exposure-response analyses of patients with IPF and other chronic fibrosing ILDs with a progressive phenotype, indicated a weak relationship between nintedanib plasma exposure and ALT and/or AST elevations. Actual administered dose might be the better predictor for the risk of developing diarrhoea of any intensity, even if plasma exposure as risk determining factor could not be ruled out (see section 4.4).
Preclinical safety data
General toxicology
Single dose toxicity studies in rats and mice indicated a low acute toxic potential of nintedanib. In repeat dose toxicology studies in young rats, irreversible changes of enamel and dentin were observed in the continuously fast-growing incisors, but not in premolars or molars. In addition, thickening of epiphyseal growth plates during bone growth phases was observed and was reversible after discontinuation. These changes are known from other VEGFR-2 inhibitors and can be considered class effects.
Diarrhoea and vomiting accompanied by reduced food consumption and loss of body weight were observed in toxicity studies in non-rodents.
There was no evidence of liver enzyme increases in rats, dogs, and cynomolgus monkeys. Mild liver enzyme increases, which were not due to serious adverse effects such as diarrhoea were only observed in rhesus monkeys.
Reproduction toxicity
In rats, embryo-foetal lethality and teratogenic effects were observed at exposure levels below human exposure at the MRHD of 150 mg twice daily. Effects on the development of the axial skeleton and on the development of the great arteries were also noted at subtherapeutic exposure levels.
In rabbits, embryo-foetal lethality and teratogenic effects were observed at an exposure approximately 3 times higher than at the MRHD but equivocal effects on the embryo-foetal development of the axial skeleton and the heart were noted already at an exposure below that at the MRHD of 150 mg twice daily.
In a pre- and postnatal development study in rats, effects on pre- and post-natal development were seen at an exposure below the MRHD.
A study of male fertility and early embryonic development up to implantation in rats did not reveal effects on the male reproductive tract and male fertility.
In rats, small amounts of radiolabelled nintedanib and/or its metabolites were excreted into the milk (≤0.5% of the administered dose).
From the 2-year carcinogenicity studies in mice and rats, there was no evidence for a carcinogenic potential of nintedanib.
Genotoxicity studies indicated no mutagenic potential for nintedanib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.