OZAWADE Film-coated tablet Ref.[27832] Active ingredients: Pitolisant
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Bioprojet Pharma, 9, rue Rameau, 75002 Paris, France Tel: +33 (0)1 47 03 66 33 Fax: +33 (0)1 47 03 66 30 e-mail: contact@bioprojet.com
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other nervous system drugs
ATC code: N07XX11
Mechanism of action
Pitolisant is an orally active histamine H3-receptor antagonist/inverse agonist which, via its blockade of histamine auto-receptors enhances the activity of brain histaminergic neurons, a major arousal system with widespread projections to the whole brain. Pitolisant also modulates various neurotransmitter systems, increasing acetylcholine, noradrenaline and dopamine release in the brain.
Clinical efficacy
The efficacy of pitolisant in the treatment of Excessive Daytime Sleepiness (EDS) in patients with Obstructive Sleep Apnoea (OSA) has been studied in two pivotal clinical studies: HAROSA I and HAROSA II.
HAROSA I studied the efficacy and safety of pitolisant in the treatment of EDS in patients with Obstructive Sleep Apnoea syndrome (OSA), and treated by Continuous Positive Airway Pressure (CPAP), but still complaining of EDS. This was a prospective, multicenter, randomized, double-blind study of pitolisant versus placebo, 12-week double-blind phase. 244 patients were analyzed (183 pitolisant, 61 placebo), 83% male, average of 53 years old, 12% over 65 years. Patients had EDS (an Epworth Sleepiness Scale [ESS] score greater than or equal to 12) and were submitted to nCPAP therapy for a minimum period of 3 months and still complaining of EDS despite the efforts made beforehand to obtain an efficient nCPAP.
The primary efficacy variable was the change in Epworth Sleepiness Scale (ESS) Score between baseline and end of treatment. During the double-blind phase, the maximum dose prescribed was 18 mg for 79.8% of the patient in the active treatment group and for 88.5% of the patients in the placebo group. The maximum dose is reached after a three-week titration, starting with 4.5 mg.
After 12 weeks DB treatment, a significant improvement of the ESS was reported with pitolisant compared to placebo (table 1).
Table 1. Overview of Efficacy results after 12 weeks in HAROSA I:
| Parameters | Treatment group (n) | Baseline score (at V2) | Final score (at V6) | Change | Difference from placebo 95% CI | P-value |
|---|---|---|---|---|---|---|
| ESS (SD) | Placebo (61) | 14.6 (2.8) | 12.1 (6.4) | -2.75 | 2.6 [-3.9, -1.4] | Ρ<0.001 |
| Pitolisant (183) | 14.9 (2.7) | 9 (4.8) | -5.52 |
Figure 1. Changes in Epworth Sleepiness Scale (ESS) score in P09-08 study Double-Blind Phase – ITT Population (N=244):
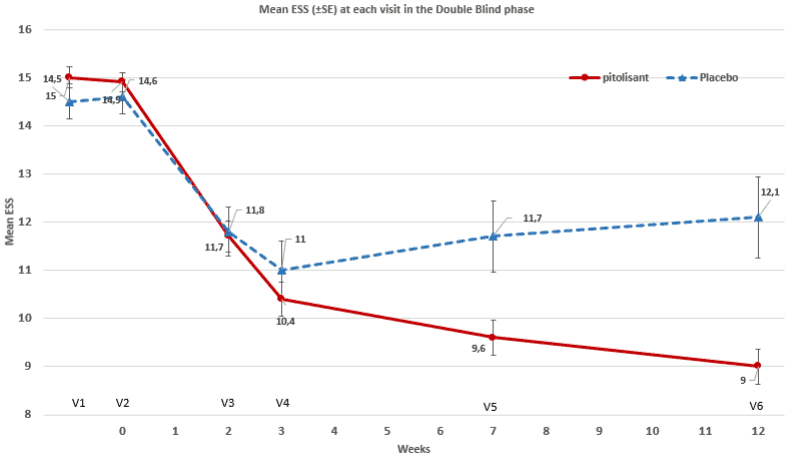
HAROSA II studied the efficacy and safety of pitolisant in the treatment of EDS in patients with Obstructive Sleep Apnoea syndrome (OSA) refusing the Continuous Positive Airway Pressure (CPAP) therapy. This was a prospective, multicenter, randomized, double-blind study of pitolisant versus placebo, 12-week double-blind phase followed by a 40-week open-label extension phase. 268 patients were analyzed (201 pitolisant, 67 placebo), 75% male, average of 52 years, 12% over 65 years. Patients had an Epworth Sleepiness Scale [ESS] score greater than or equal to 12 and were refusing to be treated by nCPAP therapy, and still complaining of EDS.
The primary efficacy variable was the change in Epworth Sleepiness Scale (ESS) score between baseline and end of treatment. During the double-blind phase, the maximum dose prescribed was 18 mg for 82.5% of the patient in the active treatment group and for 86.6% of the patients in the placebo group.
After 12 weeks DB treatment, a significant improvement of the ESS was reported with pitolisant compared to placebo (ANCOVA model adjusting for ESS and BMI at V2 and study center as random effect) (Table 2).
Table 2. Overview of Efficacy results after 12 weeks in HAROSA II:
| Parameters | Treatment group (n) | Baseline score (at V2) | Final score (at V6) | Change | Difference from placebo 95% CI | P-value |
|---|---|---|---|---|---|---|
| ESS (SD) | Placebo (67) | 15.7 (3.6) | 12.2 (6.1) | -3.6 | -2.8 [-4.0, -1.5] | Ρ<0.001 |
| Pitolisant (201) | 15.7 (3.1) | 9.1 (4.7) | -6.3 |
Figure 2. Changes in Epworth Sleepiness Scale (ESS) score in P09-09 study Double-Blind Phase – ITT Population (N=268):

In an extended analysis the two HAROSA studies were compared and combined, showing significant improvements by pitolisant compared with placebo on the main parameters (ESS, OSleR test, Pichot Fatigue Scale and CGI).
Table 3. Main efficacy results in pooled analysis HAROSA I – HAROSA II:
| Mean | 95% CI | p | |
|---|---|---|---|
| OSleR Test1 | 1.18 | 1.02, 1.35 | Ρ=0.022 |
| Pichot fatigue scale2 | -1.27 | -2.30, -0.23 | P=0.017 |
| CGI3 | -0.63 | -0.84, -0.47 | Ρ<0.001 |
Open-label data
Patients who participated in the double-blind 12 weeks period of HAROSA I and HAROSA II studies, could participate in the 40 week open-label phase. The primary objective of the open-label phase was long-term safety and effectiveness of pitolisant up to 18 mg/day. Maintenance of effect of pitolisant in EDS in OSA patients has not been established in blinded, placebo-controlled studies. In HAROSA I, 1.5% of patients discontinued study participation during the open-label phase, due to lack of efficacy and 4.0% due to adverse events. In HAROSA II, 1.3% of patients discontinued study participation during the open-label phase due to lack of efficacy and 2.5% due to adverse events.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Ozawade in all subsets of the paediatric population in Obstructive Sleep Apnoea (OSA) (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The exposure to pitolisant in healthy volunteers was assessed in studies involving more than 200 subjects that received doses of pitolisant in single administration up to 216 mg and for a duration up to 28 days.
Absorption
Pitolisant is well and rapidly absorbed with peak plasma concentration reached approximately three hours after administration. The steady-state (geometric mean, CV%) Cmax and AUC of the therapeutic dose (18 mg) is 35.5 ng/mL (59.2%) and 378 ng x h/mL (86.3%), respectively.
Upon repeated administrations, the steady state is achieved after 5-6 days of administration leading to an increased serum level around 2-fold. Inter individual variability is rather high (Geom CV% of 59.2 and 86.3 for Cmax and AUC0-24h respectively), some volunteers showing outlier high profile (without tolerance issues).
The pharmacokinetics of pitolisant is not influenced by concomitant food intake.
Distribution
Pitolisant exhibits high serum protein binding (91.4-95.2%) and demonstrates approximately equal distribution between red blood cells and plasma.
Pitolisant is widely distributed with an apparent volume of distribution of 5-10 L/kg.
Biotransformation
The metabolism of pitolisant in humans is well characterized and represents the major route of elimination. The major non-conjugated metabolites are cleaved forms of pitolisant leading to inactive major carboxylic acid metabolites, three of which being major and in a lesser extent five hydroxylated/N-oxide derivatives in several positions found in urine and serum. By combining the contribution of enzyme determined in vitro with the exposure of the main metabolites identified in the mass balance study, the estimated overall contribution of CYP enzymes in pitolisant metabolism is of 60% for CYP2D6 and of ~30% for CYP3A4/3A5 when CYP2D6 phenotype is extensive metabolizer. Several conjugated metabolites were identified, the major ones (inactive) being two glycine conjugates of carboxylic acid metabolites of pitolisant and a glucuronide of a ketone metabolite of monohydroxy desaturated pitolisant.
Inhibition/Induction
On liver microsomes, pitolisant and its major metabolites do not significantly inhibit the activities of the cytochromes CYP1A2, CYP2C9, CYP2C19, CYP2C8, CYP2B6, CYP2E1 or CYP3A4 and of uridine diphosphate glucuronosyl transferases isoforms UGT1A1, UGT1A4, UGT1A6, UGT1A9 and UGT2B7 up to the concentration of 13.3 µM, a level considerably higher than the levels achieved with therapeutic dose. Pitolisant is an inhibitor of CYP2D6 with moderate potency (IC50 = 2.6 µM).
Based on in vitro data, pitolisant and its main metabolites may induce CYP3A4 and CYP2B6 at therapeutic concentrations and by extrapolation, CYP2C, UGTs and P-gp. A clinical study was conducted to assess the effect of pitolisant on CYP3A4 and CYP2B6 using midazolam and bupropion as a CYP3A4 and a CYP2B6 model substrate, respectively. Pitolisant does not affect the pharmacokinetic of bupropion and consequently is not a CYP2B6 or a CYP1A2 inducer and should be considered a borderline/weak inducer at clinically relevant concentrations.
In vitro studies indicate that pitolisant is neither a substrate nor an inhibitor of human P-glycoprotein and breast cancer resistance protein (BCRP). Pitolisant is not a substrate of OATP1B1, OATP1B3. Pitolisant is not a significant inhibitor of OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2K at the tested concentration. Pitolisant shows greater than 50% inhibition towards OCT1 (organic cation transporters 1) at 1.33 µM, the extrapolated IC50 of pitolisant is 0.795 µM (see section 4.5).
Elimination
Pitolisant has a plasma half-life of 10-12 hours. The elimination is mainly achieved via urine (approximately 90%) through pharmacologically inactive non conjugated and glycine and glucuronide conjugated metabolites. A small fraction (2.3%) was recovered in faeces.
Linearity/non-linearity
A cross-study assessment of single-dose data shows that pitolisant exposures increase proportionally with doses between 18 and 216 mg pitolisant but slightly more than dose-proportionally over the clinical dose range of 4.5 to 18 mg.
Special populations
There are unlikely to be any clinically relevant differences in the PK of pitolisant due to sex. Pitolisant has not been studied in obese population with BMI >40 kg/m².
Elderly
In 68 to 80 years old healthy volunteers the pharmacokinetics of pitolisant is not different compared to younger patients (18 to 45 years of age). Above 80 years old, kinetics show a slight variation without clinical relevance. Limited data are available in elderly. Therefore, dosing should be adjusted according to their hepatic status (see section 4.2 and 4.4).
Renal impairment
In patients with impaired renal function (stages 2 to 4 according to the international classification of chronic kidney disease, i.e. creatinine clearance between 15 and 89 ml/min), Cmax and AUC tended to be increased by a factor of 2.5 (see section 4.2). The underlying mechanism is unknown.
Hepatic impairment
In patients with mild hepatic impairment (Child-Pugh A), AUC increased by a factor 1.4 while Cmax remained unchanged, compared with normal healthy volunteers. In patients with moderate hepatic impairment (Child-Pugh B), AUC increased by a factor 2.4, while Cmax remained unchanged (see section 4.2). Pitolisant pharmacokinetics after repeated administration in patients with hepatic impairment has not been evaluated yet.
Race
All studies have been performed mainly in Caucasians (Caucasians = 270; Black = 38; Asian = 20; Other = 3). Based on the data provided by the Applicant, the exposure appears to be similar between the different races.
CYP2D6 phenotypes and CYP3A polymorphism
The exposure to pitolisant was higher in the CYP2D6 poor metabolizers after a single dose and at steady state; Cmax and AUC(0-tau) was approximately 2.7-fold and 3.2-fold greater on Day 1 and 2.1- fold and 2.4-fold on Day 7. The serum pitolisant half-life was longer in CYP2D6 poor metabolizers compared to the extensive metabolizers.
In subjects that are CYP2D6 intermediate, extensive (normal) or ultra-rapid metabolizers, CYP2D6 is the main enzyme involved in the biotransformation of pitolisant, CYP3A is involved to a lesser extent. CYP3A4 and CYP3A5 genetic polymorphisms are unlikely to have significant effect on the pharmacokinetic of pitolisant.
In these subjects, CYP2D6 inhibitors will have an effect on the pharmacokinetic of pitolisant, not CYP3A inhibitors. In subjects that are CYP2D6 ultra-rapid metabolizers, CYP3A inducers may lead to an even more rapid elimination of pitolisant and lower exposures compared to the other subgroups. This may result in exposures below therapeutic concentrations.
In subjects that are CYP2D6 poor metabolizers or are CYP2D6 intermediate, extensive or ultra-rapid metabolizers taking CYP3A inducers, CYP3A is significantly involved in the biotransformation of pitolisant and CYP2D6 is involved to a lesser extent. Only under these conditions, genetic polymorphisms in CYP3A4 and 3A5 may have a significant effect on the pharmacokinetic of pitolisant.
In subjects that are CYP2D6 poor metabolizers, CYP3A inhibitors and inducers will have an effect on the pharmacokinetic of pitolisant and CYP2D6 inhibitors to a much lesser extent. In subjects that are CYP2D6 intermediate, extensive or ultra-rapid metabolizers taking a CYP3A inducer, a CYP3A inhibitor will lead to a decrease in the contribution of CYP3A to the overall metabolism. However, the exposure is most likely similar to that in subjects that are not taking a CYP3A inducer. Thus, in this subpopulation, CYP3A inhibition is unlikely to affect the pharmacokinetic of pitolisant.
5.3. Preclinical safety data
In rats, transient reversible convulsive episodes occurred at Tmax, that may be attributable to a metabolite abundant in this species but not in humans. In monkeys, at the highest doses, transient CNS related clinical signs including emesis, tremors and convulsions were reported. At the highest doses, rats presented some limited histopathological changes in some organs (liver, duodenum, thymus, adrenal gland and lung).
Pitolisant blocked hERG channel with an IC50 exceeding therapeutic concentrations and induced a slight QTc prolongation in dogs.
In preclinical studies, drug dependence and drug abuse liability studies were conducted in mice, rats and monkeys. However, no definitive conclusion could be drawn on tolerance, dependence and selfadministration studies.
Pitolisant was neither genotoxic nor carcinogenic.
Teratogenic effect of pitolisant was observed at maternally toxic doses (teratogenicity safety margins 7.3 and 2.6 in rats and in rabbits, respectively). At high doses, pitolisant induced sperm morphology abnormalities and decreased motility without any significant effect on fertility indexes in male rats and it decreased the percentage of live conceptuses and increased post-implantation loss in female rats (safety margin of 2.3). It caused a delay in post-natal development (safety margin of 2.3). Pitolisant/metabolites were shown to cross the placenta barrier and secreted in breast milk in animals.
Juvenile toxicity studies
Juvenile toxicity studies in rats revealed that the administration of pitolisant at high doses induced a dose related mortality and convulsive episode that may be attributable to a metabolite abundant in rats but not in humans.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.