PHESGO Solution for injection Ref.[51368] Active ingredients: Pertuzumab Pertuzumab and Trastuzumab Trastuzumab
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies
ATC code: L01XY02
Mechanism of action
Phesgo contains pertuzumab and trastuzumab which provides the therapeutic effect of this medicinal product and vorhyaluronidase alfa, an enzyme used to increase the dispersion and absorption of coformulated substances when administered subcutaneously.
Pertuzumab and trastuzumab are recombinant humanised IgG1 monoclonal antibodies which target the human epidermal growth factor receptor 2 (HER2). Both substances bind to distinct HER2 subdomains without competing and have complementary mechanisms for disrupting HER2 signalling:
- Pertuzumab specifically targets the extracellular dimerization domain (subdomain II) of HER2 and thereby blocks ligand-dependent heterodimerization of HER2 with other HER family members, including epidermal growth factor receptor (EGFR), HER3 and HER4. As a result, pertuzumab inhibits ligand-activated intracellular signalling through two major signalling pathways, mitogen-activated protein (MAP) kinase and phosphoinositide 3-kinase (PI3K). Inhibition of these signalling pathways can result in cell growth arrest and apoptosis, respectively
- Trastuzumab binds to sub-domain IV, of the extracellular domain of the HER2 protein to inhibit the ligand-independent, HER2 mediated proliferation and survival signals in human tumour cells that over express HER2.
Additionally, both substances mediate antibody-dependent cell-mediated cytotoxicity (ADCC). In vitro, both pertuzumab and trastuzumab ADCC are exerted preferentially on HER2-overexpressing cancer cells compared with cancer cells that do not overexpress HER2.
Clinical efficacy and safety
This section is presenting the clinical experience from Phesgo fixed dose combination of pertuzumab and trastuzumab and from intravenous pertuzumab in combination with trastuzumab in patients with HER2 overexpressing early and metastatic breast cancer.
Clinical experience of Phesgo in patients with HER2 positive early breast cancer
The clinical experience of Phesgo is based on data from a Phase III clinical trial (FEDERICA WO40324) and a Phase II clinical trial (PHRANCESCA MO40628) in patients with HER2 overexpressing early breast cancer. HER2 overexpression was determined at a central laboratory and defined as a score of 3+ by IHC or an ISH amplification ratio ≥2.0 in the trial outlined below.
FEDERICA (WO40324)
FEDERICA is an open-label, multicenter, randomized study conducted in 500 patients with HER2- positive early breast cancer that is operable or locally advanced (including inflammatory) with a tumour size >2 cm or node-positive in the neoadjuvant and adjuvant setting. Patients were randomized to receive 8 cycles of neoadjuvant chemotherapy with concurrent administration of 4 cycles of either Phesgo or intravenous pertuzumab and trastuzumab during cycles 5-8. Investigators selected one of the two following neoadjuvant chemotherapy for individual patients:
- 4 cycles of doxorubicin (60 mg/m²) and cyclophosphamide (600 mg/m²) every 2 weeks followed by paclitaxel (80 mg/m²) weekly for 12 weeks
- 4 cycles of doxorubicin (60 mg/m²) and cyclophosphamide (600 mg/m²) every 3 weeks followed by 4 cycles of docetaxel (75 mg/m² for the first cycle and then 100 mg/m² at subsequent cycles at the investigator's discretion) every 3 weeks
Following surgery, patients continued therapy with Phesgo or intravenous pertuzumab and trastuzumab as treated prior to surgery for an additional 14 cycles, to complete 18 cycles of HER2 targeted therapy. Patients also received adjuvant radiotherapy and endocrine therapy as per local practice. In adjuvant period, substitution of intravenous trastuzumab for subcutaneous trastuzumab was permitted at investigator discretion. HER2-targeted therapy was administered every 3 weeks according to Table 3 as follows:
Table 3. Dosing and administration of Phesgo, intravenous pertuzumab, intravenous trastuzumab, and subcutaneous trastuzumab:
| Medicinal Products | Administration | Dose | |
|---|---|---|---|
| Loading | Maintenance | ||
| Phesgo | Subcutaneous injection | 1200 mg/600 mg | 600 mg/600 mg |
| Pertuzumab | Intravenous infusion | 840 mg | 420 mg |
| Trastuzumab | Intravenous infusion | 8 mg/kg | 6 mg/kg |
| Trastuzumab | Subcutaneous injection | 600 mg | |
FEDERICA was designed to demonstrate non-inferiority of the pertuzumab Cycle 7 (i.e., pre-dose Cycle 8) serum Ctrough of pertuzumab within Phesgo compared with intravenous pertuzumab (primary endpoint). Additional secondary endpoints included non-inferiority of the Cycle 7 serum trastuzumab Ctrough of trastuzumab within Phesgo compared with intravenous trastuzumab, efficacy (locally assessed total pathological complete response, tpCR), and safety outcomes. Demographics were well balanced between the two treatment arms and the median age of patients treated in the study was 51 years. The majority of patients had hormone receptor-positive disease (61.2%), node-positive disease (57.6%), and were Caucasian (65.8%).
For non-inferiority of the pertuzumab and trastuzumab exposures from Phesgo refer to section 5.2. For safety profile refer to section 4.8.
The analysis of secondary efficacy endpoint, tpCR (locally assessed), defined as an absence of invasive disease in the breast and axilla (ypT0/is, ypN0) is shown in Table 4.
Table 4. Summary of total pathological complete response (tpCR):
| Phesgo (n= 248) | Intravenous pertuzumab + trastuzumab (n= 252) | |
|---|---|---|
| tpCR (ypT0/is, ypN0) | 148 (59.7%) | 150 (59.5%) |
| Exact 95% CI for tpCR rate1 | (53.28; 65.84) | (53.18; 65.64) |
| Difference in tpCR rate (subcutaneous minus intravenous arm) | 0.15 | |
| 95% CI for the difference in tpCR2 rate | -8.67 to 8.97 | |
1 Confidence interval for one sample binomial using Pearson-Clopper method
2 Hauck-Anderson continuity correction has been used in this calculation
PHRANCESCA (MO40628)
Study MO40628 investigated the safety of switching between intravenous pertuzumab and trastuzumab and Phesgo subcutaneous and vice versa (see section 4.8) with a primary objective to evaluate patient preference for either the intravenous or the subcutaneous route of administration: 85% of patients preferred the subcutaneous route, whereas 13.8% preferred the IV administration, and 1.2% had no preference. A total of 160 patients were included in this 2-arm, cross-over study: 80 patients were randomized to Arm A (3 cycles of intravenous pertuzumab and trastuzumab followed by 3 cycles of Phesgo) and 80 patients were randomized to Arm B (3 cycles of Phesgo followed by 3 cycles intravenous pertuzumab and trastuzumab). At primary analysis, the median exposure to adjuvant pertuzumab and trastuzumab (both IV and SC administration) was 11 cycles (range: 6 to 15).
Clinical experience of intravenous pertuzumab in combination with trastuzumab in HER2 positive breast cancer
The clinical experience of intravenous pertuzumab in combination with trastuzumab is based on data from two randomised neoadjuvant phase II trials in early breast cancer (one controlled), a nonrandomised neoadjuvant phase II trial, a randomised phase III trial in the adjuvant setting and a randomised phase III trial and a single-arm phase II trial in metastatic breast cancer. HER2 overexpression was determined at a central laboratory and defined as a score of 3+ by IHC or an ISH amplification ratio ≥2.0 in the trials outlined below.
Early breast cancer
Neoadjuvant treatment
In the neoadjuvant setting, locally advanced and inflammatory breast cancers are considered as highrisk irrespective of hormone receptor status. In early stage breast cancer, tumour size, grade, hormone receptor status and lymph node metastases should be taken into account in the risk assessment.
The indication in the neoadjuvant treatment of breast cancer is based on demonstration of an improvement in pathological complete response rate, and trends to improvement in disease-free survival (DFS) that nevertheless do not establish or precisely measure a benefit with regard to longterm outcomes, such as overall survival (OS) or DFS.
NEOSPHERE (WO20697)
NEOSPHERE is a phase II, multicentre, multinational randomised controlled trial with pertuzumab and was conducted in 417 adult female patients with newly diagnosed, early, inflammatory or locally advanced HER2-positive breast cancer (T2-4d; primary tumour >2 cm in diameter) who had not received prior trastuzumab, chemotherapy or radiotherapy. Patients with metastases, bilateral breast cancer, clinically important cardiac risk factors (see section 4.4) or LVEF <55% were not included. The majority of patients were less than 65 years old.
Patients were randomised to receive one of the following neoadjuvant regimens for 4 cycles prior to surgery:
- Trastuzumab plus docetaxel
- Pertuzumab plus trastuzumab and docetaxel
- Pertuzumab plus trastuzumab
- Pertuzumab plus docetaxel.
Randomisation was stratified by breast cancer type (operable, locally advanced, or inflammatory) and oestrogen receptor (ER) or progesterone (PgR) positivity.
Pertuzumab was given intravenously at an initial dose of 840 mg, followed by 420 mg every three weeks. Trastuzumab was given intravenously at an initial dose of 8 mg/kg, followed by 6 mg/kg every three weeks. Docetaxel was given intravenously at an initial dose of 75 mg/m² followed by 75 mg/m² or 100 mg/m² (if tolerated) every 3 weeks. Following surgery all patients received 3 cycles of 5-fluorouracil (600 mg/m²), epirubicin (90 mg/m²), cyclophosphamide (600 mg/m²) (FEC) given intravenously every three weeks, and trastuzumab administered intravenously every three weeks to complete one year of therapy. Patients who only received pertuzumab plus trastuzumab prior to surgery subsequently received both FEC and docetaxel post-surgery.
The primary endpoint of the study was pathological complete response (pCR) rate in the breast (ypT0/is). Secondary efficacy endpoints were clinical response rate, breast conserving surgery rate (T2-3 tumours only), DFS, and progression-free survival (PFS). Additional exploratory pCR rates included nodal status (ypT0/isN0 and ypT0N0).
Demographics were well balanced (median age was 49-50 years, the majority were caucasian (71%)) and all patients were female. Overall 7% of patients had inflammatory breast cancer, 32% had locally advanced breast cancer and 61% had operable breast cancer. Approximately half the patients in each treatment group had hormone receptor-positive disease (defined as ER positive and/or PgR positive).
The efficacy results are presented in Table 5. A statistically significant improvement in pCR rate (ypT0/is) was observed in patients receiving pertuzumab plus trastuzumab and docetaxel compared to patients receiving trastuzumab and docetaxel (45.8% vs. 29.0%, p value= 0.0141). A consistent pattern of results was observed regardless of pCR definition. The difference in pCR rate is considered likely to translate into a clinically meaningful difference in long term outcomes and is supported by positive trends in PFS (hazard ratio [HR] = 0.69; 95% CI 0.34; 1.40) and DFS (HR = 0.60; 95% CI 0.28; 1.27).
The pCR rates as well as the magnitude of benefit with pertuzumab (pertuzumab plus trastuzumab and docetaxel compared to patients receiving trastuzumab and docetaxel) were lower in the subgroup of patients with hormone receptor-positive tumours (difference of 6% in pCR in the breast) than in patients with hormone receptor-negative tumours (difference of 26.4% in pCR in the breast). pCR rates were similar in patients with operable versus locally advanced disease. There were too few patients with inflammatory breast cancer to draw any firm conclusions but the pCR rate was higher in patients who received pertuzumab plus trastuzumab and docetaxel.
TRYPHAENA (BO22280)
TRYPHAENA is a multicentre, randomised phase II clinical trial conducted in 225 adult female patients with HER2-positive locally advanced, operable, or inflammatory breast cancer (T2-4d; primary tumour >2 cm in diameter) who had not received prior trastuzumab, chemotherapy or radiotherapy. Patients with metastases, bilateral breast cancer, clinically important cardiac risk factors (see section 4.4) or LVEF <55% were not included. The majority of patients were less than 65 years old. Patients were randomised to receive one of three neoadjuvant regimens prior to surgery as follows:
- 3 cycles of FEC followed by 3 cycles of docetaxel, all given concurrently with pertuzumab and trastuzumab
- 3 cycles of FEC alone followed by 3 cycles of docetaxel, with trastuzumab and pertuzumab given concurrently
- 6 cycles of TCH in combination with pertuzumab.
Randomisation was stratified by breast cancer type (operable, locally advanced, or inflammatory) and ER and/or PgR positivity.
Pertuzumab was given intravenously at an initial dose of 840 mg, followed by 420 mg every three weeks. Trastuzumab was given intravenously at an initial dose of 8 mg/kg, followed by 6 mg/kg every three weeks. FEC (5-fluorouracil [500 mg/m²], epirubicin [100 mg/m²], cyclophosphamide [600 mg/m²]) were given intravenously every three weeks for 3 cycles. Docetaxel was given as an initial dose of 75 mg/m² intravenous infusion every three weeks with the option to escalate to 100 mg/m² at the investigator's discretion if the initial dose was well tolerated. However, in the group treated with pertuzumab in combination with TCH, docetaxel was given intravenously at 75 mg/m² (no escalation was permitted) and carboplatin (AUC 6) was given intravenously every three weeks. Following surgery all patients received trastuzumab to complete one year of therapy.
The primary endpoint of this study was cardiac safety during the neoadjuvant treatment period of the study. Secondary efficacy endpoints were pCR rate in the breast (ypT0/is), DFS, PFS and OS.
Demographics were well balanced between arms (median age was 49-50 years, the majority were Caucasian [77%]) and all patients were female. Overall 6% of patients had inflammatory breast cancer, 25% had locally advanced breast cancer and 69% had operable breast cancer. Approximately half the patients in each treatment group had ER-positive and/or PgR-positive disease.
Compared with published data for similar regimens without pertuzumab, high pCR rates were observed in all 3 treatment arms (see Table 5). A consistent pattern of results was observed regardless of pCR definition used. The pCR rates were lower in the subgroup of patients with hormone receptorpositive tumours (range 46.2% to 50.0%) than in patients with hormone receptor-negative tumours (range 65.0% to 83.8%).
pCR rates were similar in patients with operable and locally advanced disease. There were too few patients with inflammatory breast cancer to draw any firm conclusions.
Table 5. NEOSPHERE (WO20697) and TRYPHAENA (BO22280): Overview of efficacy (Intent to treat population):
| NEOSPHERE (WO20697) | TRYPHAENA (BO22280) | ||||||
|---|---|---|---|---|---|---|---|
| Parameter | Trastuzumab + docetaxel N=107 | Pertuzumab + trastuzumab + docetaxel N=107 | Pertuzumab + trastuzumab N=107 | Pertuzumab + docetaxel N=96 | Pertuzumab + trastuzumab + FEC → pertuzumab + trastuzumab + docetaxel N=73 | FEC → Pertuzumab + trastuzumab + docetaxel N=75 | Pertuzumab + TCH N=77 |
| pCR rate in the breast (ypT0/is) n (%) [95% CI]1 | 31 (29,0%) [20,6, 38,5] | 49 (45,8%) [36,1, 55,7] | 18 (16,8%) [10,3, 25,3] | 23 (24,0%) [15,8, 33,7] | 45 (61,6%) [49,5, 72,8] | 43 (57,3%) [45,4, 68,7] | 51 (66,2%) [54,6, 76,6] |
| Difference in pCR rates2 [95% CI]3 | +16,8% [3,5, 30,1] | -12,2% [-23,8, -0,5] | -21,8% [-35,1, -8,5] | NA | NA | NA | |
| p-value (with Simes corr. for CMH test)4 | 0.0141 (vs. trastuzumab + docetaxel) | 0.0198 (vs. trastuzumab + docetaxel) | 0.0030 (vs. pertuzumab + trastuzumab + docetaxel) | NA | NA | NA | |
| pCR rate in the breast and lymph node (ypT0/is N0) n (%) [95% CI] | 23 (21,5%) [14,1, 30,5] | 42 (39,3%) [30,3, 49,2] | 12 (11,2%) [5,9, 18,8] | 17 (17,7%) [10,7, 26,8] | 41 (56,2%) [44,1, 67,8] | 41 (54,7%) [42,7, 66,2] | 49 (63,6%) [51,9, 74,3] |
| ypT0 N0 n (%) [95% CI] | 13 (12,1%) [6,6, 19,9] | 35 (32,7%) [24,0, 42,5] | 6 (5,6%) [2,1, 11,8] | 13 (13,2%) [7,4, 22,0] | 37 (50,7%) [38,7, 62,6] | 34 (45,3%) [33,8, 57,3] | 40 (51,9%) [40,3, 63,5] |
| Clinical Response5 | 79 (79,8%) | 89 (88,1%) | 69 (67,6%) | 65 (71,4%) | 67 (91,8%) | 71 (94,7%) | 69 (89,6%) |
FEC: 5-fluorouracil, epirubicin, cyclophosphamide; TCH: docetaxel, carboplatin and trastuzumab, CMH: Cochran–Mantel–Haenszel
1 95% CI for one sample binomial using Pearson-Clopper method.
2 Treatment pertuzumab+trastuzumab+docetaxel and pertuzumab+trastuzumab are compared to Trastuzumab+Docetaxel while pertuzumab+docetaxel is compared to pertuzumab+trastuzumab+docetaxel.
3 Approximate 95 % CI for difference of two response rates using Hauck-Anderson method.
4 p-value from Cochran-Mantel-Haenszel test, with Simes multiplicity adjustment.
5 Clinical response represents patients with a best overall response of CR or PR during the neoadjuvant period (in the primary breast lesion).
BERENICE (WO29217)
BERENICE is a non-randomized, open-label, multicentre, multinational, Phase II trial conducted in 401 patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (with primary tumours >2 cm in diameter or node-positive disease).
The BERENICE study included two parallel groups of patients. Patients considered suitable for neoadjuvant treatment with trastuzumab plus anthracycline/taxane-based chemotherapy were allocated to receive one of the two following regimens prior to surgery as follows:
- Cohort A - 4 cycles of two weekly dose-dense doxorubicin and cyclophosphamide followed by 4 cycles of pertuzumab in combination with trastuzumab and paclitaxel.
- Cohort B - 4 cycles of FEC followed by 4 cycles of pertuzumab in combination with trastuzumab and docetaxel.
Following surgery all patients received pertuzumab and trastuzumab intravenously every 3 weeks to complete 1 year of therapy.
The primary endpoint of the BERENICE trial is cardiac safety in the neoadjuvant period of the trial. The primary endpoint of cardiac safety, i.e. the incidence of NYHA Class III/IV LVD and LVEF declines, was consistent with previous data in the neoadjuvant setting (see sections 4.4. and 4.8).
Adjuvant treatment
In the adjuvant setting, based on data from the APHINITY study, HER2-positive early breast cancer patients at high risk of recurrence are defined as those with lymph node-positive or hormone receptornegative disease.
APHINITY (BO25126)
APHINITY is a multicentre, randomised, double-blind, placebo-controlled Phase III trial conducted in 4804 patients with HER2-positive early breast cancer who had their primary tumour excised prior to randomisation. Patients were then randomised to receive pertuzumab or placebo, in combination with adjuvant trastuzumab and chemotherapy. Investigators selected one of the following anthracyclinebased or non-anthracycline-based chemotherapy regimens for individual patients:
- 3 or 4 cycles of FEC or 5-fluorouracil, doxorubicin and cyclophosphamide (FAC), followed by 3 or 4 cycles of docetaxel or 12 cycles of weekly paclitaxel
- 4 cycles of AC or epirubicin and cyclophosphamide (EC), followed by 3 or 4 cycles of docetaxel or 12 cycles of weekly paclitaxel
- 6 cycles of docetaxel in combination with carboplatin
Pertuzumab and trastuzumab were administered intravenously (see section 4.2) every 3 weeks starting on Day 1 of the first taxane-containing cycle, for a total of 52 weeks (up to 18 cycles) or until recurrence, withdrawal of consent or unmanageable toxicity. Standard doses of 5-fluorouracil, epirubicin, doxorubicin, cyclophosphamide, docetaxel, paclitaxel and carboplatin were administered. After completion of chemotherapy, patients received radiotherapy and/or hormone therapy as per local clinical standard.
The primary endpoint of the study was invasive disease-free survival (IDFS), defined as the time from randomisation to first occurrence of ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause. Secondary efficacy endpoints were IDFS including second primary non-breast cancer, OS, DFS, recurrence-free interval (RFI) and distant recurrence-free interval (DRFI).
Demographics were well balanced between the two treatment arms. The median age was 51 years, and over 99% of patients were female. The majority of patients had node-positive (63%) and/or hormone receptor-positive disease (64%), and were Caucasian (71%). After a median follow-up of 45.4 months, the APHINITY study showed a 19% (HR = 0.81; 95% CI 0.66; 1.00 p-value 0.0446) reduction in risk of recurrence or death in patients randomised to receive pertuzumab compared with patients randomised to receive placebo.
The efficacy results from the APHINITY trial are summarised in Table 6 and in Figure 1.
Table 6. Overall efficacy: Intent to treat population:
| Pertuzumab + trastuzumab + chemotherapy N=2.400 | Placebo + trastuzumab + chemotherapy N=2.404 | |
|---|---|---|
| Primary Endpoint | ||
| Invasive disease free survival (IDFS) | ||
| Number (%) of patients with event | 171 (7.1%) | 210 (8.7%) |
| HR [95% CI] | 0.81 [0.66, 1.00] | |
| p-value (Log-Rank test, stratified1) | 0.0446 | |
| 3 year event-free rate2 95% CI] | 94.1 [93.1, 95.0] | 93.2 [92.2, 94.3] |
| Secondary Endpoint1 | ||
| IDFS including second primary non-breast cancer | ||
| Number (%) patients with event | 189 (7.9%) | 230 (9.6%) |
| HR [95% CI] | 0.82 [0.68, 0.99] | |
| p-value (Log-Rank test, stratified1) | 0.0430 | |
| 3 year event-free rate2 [95% CI] | 93.5 [92.5, 94.5] | 92.5 [91.4, 93.6] |
| Disease free survival (DFS) | ||
| Number (%) patients with event | 192 (8.0%) | 236 (9.8%) |
| HR [95% CI] | 0.81 [0.67, 0.98] | |
| p-value (Log-Rank test, stratified1) | 0.0327 | |
| 3 year event-free rate2 [95% CI] | 93.4 [92.4, 94.4] | 92.3 [91.2, 93.4] |
| Overall survival (OS)3 | ||
| Number (%) patients with event | 80 (3.3%) | 89 (3.7%) |
| HR [95% CI] | 0.89 [0.66, 1.21] | |
| p-value (Log-Rank test, stratified1) | 0.4673 | |
| 3 year event-free rate2 [95% CI] | 97.7 [97.0, 98.3] | 97.7 [97.1, 98.3] |
Key to abbreviations (Table 6): HR: Hazard Ratio; CI: Confidence Interval
1 All analyses stratified by nodal status, protocol version, central hormone receptor status, and adjuvant chemotherapy regimen.
2 3-year event-free rate derived from Kaplan-Meier estimates.
3 Data from first interim analysis.
Figure 1. Kaplan-Meier curve of invasive disease free survival:
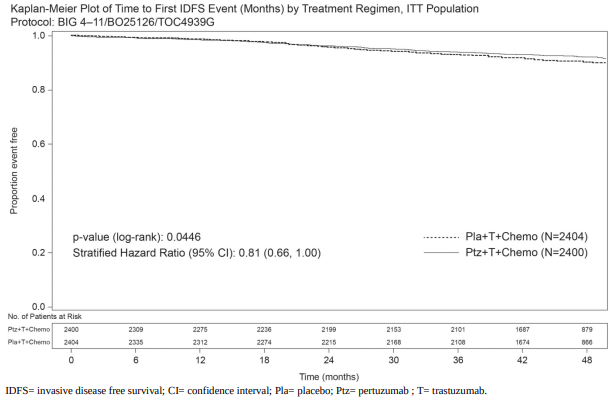
The estimate of IDFS at 4-years was 92.3% in the pertuzumab-treated group versus 90.6% in the placebo-treated group. At the time of the estimate the median follow-up was 45.4 months.
Results of subgroup analysis
At the time of the primary analysis, the benefits of pertuzumab were more apparent in subgroups of patients a high risk of recurrence: patients with node-positive or hormone receptor-negative disease (see table 7).
Table 7. Efficacy results in subgroups by nodal status and hormone receptor status1:
| Population | Number of IDFS events/Total N (%) | Unstratified HR (95% CI) | |
|---|---|---|---|
| Pertuzumab + trastuzumab + chemotherapy | Placebo + trastuzumab + chemotherapy | ||
| Nodal status | |||
| Positive | 139/1.503 (9,2%) | 181/1.502 (12,1%) | 0,77 (0,62, 0,96) |
| Negative | 32/897 (3,6%) | 29/902 (3,2%) | 1,13 (0,68, 1,86) |
| Hormone receptor status | |||
| Negative | 71/864 (8,2%) | 91/858 (10,6%) | 0,76 (0,56, 1,04) |
| Positive | 100/1536 (6,5%) | 119/1.546 (7,7%) | 0,86 (0,66, 1,13) |
1 Prespecified subgroup analyses without adjusting for multiple comparisons, therefore, results are considered descriptive.
Estimates of IDFS rates in the lymph node-positive subgroup were 92.0% versus 90.2% at 3 years and 89.9% vs. 86.7% at 4 years in pertuzumab-treated patients versus placebo-treated patients, respectively. In the lymph node-negative subgroup, estimates of IDFS rates were 97.5% versus 98.4% at 3 years and 96.2% versus 96.7% at 4 years in pertuzumab-treated patients versus placebotreated patients, respectively. In the hormone receptor-negative subgroup, estimates of IDFS rates were 92.8% versus 91.2% at 3 years and 91.0% versus 88.7% at 4 years in pertuzumab-treated patients versus placebo-treated patients, respectively. In the hormone receptor-positive subgroup estimates of IDFS rates were 94.8% versus 94.4% at 3 years and 93.0% versus 91.6% at 4 years in pertuzumab-treated patients versus placebo-treated patients, respectively.
Patient Reported Outcomes (PRO)
Secondary endpoints included the assessment of patient-reported global health status, role and physical function, and treatment symptoms using the EORTC QLQ-C30 and EORTC QLQ-BR23 questionnaires. In the analyses of patient-reported outcomes, a 10-point difference was considered clinically meaningful.
Patients' physical function, global health status and diarrhoea scores showed a clinically meaningful change during chemotherapy in both treatment arms. The mean decrease from baseline at that time for physical function was -10.7 (95% CI -11.4; -10.0) in the pertuzumab arm and 10.6 (95% CI 11.4; -9.9) in the placebo arm; global health status was -11.2 (95% CI -12.2; -10.2) in the pertuzumab arm and -10.2 (95% CI -11.1; -9.2) in the placebo arm. Change in diarrhoea symptoms increased to +22.3 (95% CI 21.0; 23.6) in the pertuzumab arm versus +9.2 (95% CI 8.2; 10.2) in the placebo arm.
Thereafter in both arms physical function and global health status scores returned to baseline levels during targeted treatment. Diarrhoea symptoms returned to baseline after HER2 therapy in the pertuzumab arm. The addition of pertuzumab to trastuzumab plus chemotherapy did not affect patients' overall role function over the course of the study.
Metastatic breast cancer
Pertuzumab in combination with trastuzumab and docetaxel
CLEOPATRA (WO20698) is a multicentre, randomised, double-blind, placebo-controlled phase III clinical trial conducted in 808 patients with HER2-positive metastatic or locally recurrent unresectable breast cancer. Patients with clinically important cardiac risk factors were not included (see section 4.4). Due to the exclusion of patients with brain metastases no data are available on pertuzumab activity on brain metastases. There is very limited data available in patients with unresectable locally recurrent disease. Patients were randomised 1:1 to receive placebo + trastuzumab + docetaxel or pertuzumab + trastuzumab + docetaxel.
Pertuzumab and trastuzumab were given at standard doses in a 3-weekly regimen. Patients were treated with pertuzumab and trastuzumab until disease progression, withdrawal of consent or unmanageable toxicity. Docetaxel was given as an initial dose of 75 mg/m² as an intravenous infusion every three weeks for at least 6 cycles. The dose of docetaxel could be escalated to 100 mg/m² at the investigator's discretion if the initial dose was well tolerated.
The primary endpoint of the study was PFS as assessed by an independent review facility (IRF) and defined as the time from the date of randomisation to the date of disease progression or death (from any cause) if the death occurred within 18 weeks of the last tumour assessment. Secondary efficacy endpoints were OS, PFS (investigator-assessed), objective response rate (ORR), duration of response, and time to symptom progression according to the FACT B Quality of Life questionnaire.
Approximately half the patients in each treatment group had hormone receptor-positive disease (defined as ER-positive and/or PgR-positive) and approximately half of the patients in each treatment group had received prior adjuvant or neoadjuvant therapy. Most of these patients had received prior anthracycline therapy and 11% of all patients had received prior trastuzumab. A total of 43% of patients in both treatment groups had previously received radiotherapy. Patients' median LVEF at baseline was 65.0% (range 50%-88%) in both groups.
The efficacy results from the CLEOPATRA study are summarised in Table 8. A statistically significant improvement in IRF-assessed PFS was demonstrated in the pertuzumab-treated group compared with the placebo-treated group. The results for investigator-assessed PFS were similar to those observed for IRF-assessed PFS.
Table 8. Summary of efficacy from CLEOPATRA study:
| Parameter | Placebo + trastuzumab + docetaxel n=406 | Pertuzumab + trastuzumab + docetaxel n=402 | HR (95% CI) | p-value |
|---|---|---|---|---|
| Progression-free furvival (independent review) - primary endpoint* | ||||
| no. of patients with an event Median months | 242 (59%) 12,4 | 191 (47,5%) 18,5 | 0,62 [0,51, 0,75] | <0,0001 |
| Overall survival - secondary endpoint** | ||||
| no. of patients with an event Median months | 221 (54,4%) 40,8 | 168 (41,8%) 56,5 | 0,68 [0,56, 0,84] | 0,0002 |
| Objective response rate (ORR)^ - secondary endpoint | ||||
| no. of patients with measurable disease Responders*** 95 % CI for ORR Complete response (CR) Partial response (PR) Stable disease (SD) Progressive disease (PD) | 336 233 (69,3%) [64,1, 74,2] 14 (4,2%) 219 (65,2%) 70 (20,8%) 28 (8,3%) | 343 275 (80,2%) [75,6, 84,3] 19 (5,5%) 256 (74,6%) 50 (14,6%) 13 (3,8%) | Difference in 10,8% [4,2, 17,5] | 0,0011 |
| Duration of response†^ | ||||
| n= Median weeks 95% CI for median | 233 54,1 [46, 64] | 275 87,6 [71, 106] | ||
* Primary progression-free survival analysis, cutoff date 13th May 2011.
** Event-driven final overall survival, cutoff date 11th February 2014.
*** Patients with best overall response of confirmed CR or PR by RECIST.
† Evaluated in patients with best overall response of CR or PR.
^ Objective response rate and duration of response are based on IRF-assessed tumour assessments.
Consistent results were observed across pre-specified patient subgroups including the subgroups based on stratification factors of geographic region and prior adjuvant/neoadjuvant therapy or de novo metastatic breast cancer (see Figure 2). A post hoc exploratory analysis revealed that for patients who had received prior trastuzumab (n=88), the hazard ratio for IRF-assessed PFS was 0.62 (95% CI 0.35; 1.07), compared with 0.60 (95% CI 0.43; 0.83) for patients who had received prior therapy which did not include trastuzumab (n= 288).
Figure 2. IRF-assessed PFS by patient subgroup:

The event-driven final analysis of OS was performed when 389 patients had died (221 in the placebotreated group and 168 in the pertuzumab-treated group). The statistically significant OS benefit in favour of the pertuzumab-treated group, previously observed at an interim analysis of OS (performed one year after the primary analysis), was maintained (HR = 0.68; p=0.0002 log-rank test). The median time to death was 40.8 months in the placebo-treated group and 56.5 months in the pertuzumab-treated group (see Table 8, Figure 3).
A descriptive analysis of OS performed at the end of the study when 515 patients had died (280 in the placebo-treated group and 235 in the pertuzumab-treated group) showed that the statistically significant OS benefit in favour of the pertuzumab-treated group was maintained over time after a median follow-up of 99 months (HR = 0.69; p <0.0001 log-rank test; median time to death 40.8 months [placebo-treated group] versus 57.1 months [pertuzumab-treated group]). Landmark survival estimates at 8 years were 37% in the pertuzumab-treated group and 23% in the placebotreated group.
Figure 3. Kaplan-Meier curve of event-driven overall survival:

No statistically significant differences were found between the two treatment groups in Health Related Quality of Life as assessed by FACT-B TOI-PFB scores.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Phesgo in all subsets of the paediatric population in breast cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The PK results for the primary endpoint of pertuzumab Cycle 7 Ctrough (i.e., pre-dose cycle 8), showed non-inferiority of pertuzumab within Phesgo (geometric mean 88.7 mcg/mL) compared to intravenous pertuzumab (geometric mean 72.4 mcg/mL) with a geometric mean ratio of 1.22 (90% CI: 1.14-1.31). The lower boundary of the two-sided 90% confidence interval for the geometric mean ratio of pertuzumab within Phesgo and intravenous pertuzumab was 1.14, i.e., greater than the predefined margin of 0.8.
The PK results for the secondary endpoint, trastuzumab Cycle 7 Ctrough (i.e., predose Cycle 8), showed non-inferiority of trastuzumab within Phesgo (geometric mean 57.5 mcg/mL) compared to intravenous trastuzumab (geometric mean 43.2 mcg/mL) with a geometric mean ratio of 1.33 (90% CI: 1.24-1.43).
Absorption
The median maximum serum concentration (Cmax) of pertuzumab within Phesgo and time to maximal concentration (Tmax) were 157 mcg/mL and 3.82 days, respectively. Based on population PK analysis, the absolute bioavailability was 0.712 and the first-order absorption rate (Ka) is 0.348 (1/day).
The median Cmax of trastuzumab within Phesgo and Tmax were 114 mcg/mL and 3.84 days, respectively. Based on population PK analysis, the absolute bioavailability was 0.771 and the Ka is 0.404 (1/day).
Distribution
Based on population PK analysis, the volume of distribution of the central (Vc) compartment of pertuzumab within Phesgo in the typical patient, was 2.77 litres.
Based on population PK analysis, the Vc compartment of subcutaneous trastuzumab in the typical patient, was 2.91 litres.
Biotransformation
The metabolism of Phesgo has not been directly studied. Antibodies are cleared principally by catabolism.
Elimination
Based on population PK analysis, the clearance of pertuzumab within Phesgo was 0.163 L/day and the elimination half-life (t½) was approximately 24.3 days.
Based on population PK analysis, the clearance of trastuzumab within Phesgo was 0.111 L/day. Trastuzumab is estimated to reach concentrations that are <1 mcg/mL (approximately 3% of the population predicted Cmin,ss, or about 97% washout) in at least 95% patients 7 months after the last dose.
Elderly patients
No studies have been conducted to investigate the pharmacokinetics of Phesgo in elderly patients.
In population PK analyses of pertuzumab within Phesgo and intravenous pertuzumab, age was not found to significantly affect PK of pertuzumab.
In population PK analyses of subcutaneous or intravenous trastuzumab, age has been shown to have no effect on the disposition of trastuzumab.
Renal impairment
No studies have been conducted to investigate the pharmacokinetics of Phesgo in patients with renal impairment.
Based on population PK analyses of pertuzumab within Phesgo and intravenous pertuzumab, renal impairment was shown not to affect pertuzumab exposure; however, only limited data from patients with severe renal impairment were included in population pharmacokinetic analyses.
In a population PK analysis of subcutaneous and intravenous trastuzumab, renal impairment was shown not to affect trastuzumab disposition.
Hepatic impairment
No formal PK study has been conducted in patients with hepatic impairment. Based on population PK analyses of pertuzumab within Phesgo, mild hepatic impairment was shown not to affect pertuzumab exposure. However, only limited data from patients with mild hepatic impairment were included in population PK analyses. IgG1 molecules such as pertuzumab and trastuzumab are catabolised by widely distributed proteolytic enzymes not restricted to hepatic tissue. Therefore, changes in hepatic function are unlikely to have an effect on the elimination of pertuzumab and trastuzumab.
5.3. Preclinical safety data
No dedicated studies were conducted with the combination of subcutaneous pertuzumab, trastuzumab, and vorhyaluronidase alfa.
Pertuzumab
No specific fertility studies in animals have been performed to evaluate the effect of pertuzumab. No definitive conclusion on adverse effects can be drawn on the male reproductive organs in cynomolgus monkey repeated dose toxicity.
Reproductive toxicology studies have been conducted in pregnant cynomolgus monkeys (Gestational Day (GD) 19 through to GD 50) at initial doses of 30 to 150 mg/kg followed by bi weekly doses of 10 to 100 mg/kg. These dose levels resulted in clinically relevant exposures of 2.5 to 20-fold greater than the recommended human subcutaneous dose, based on Cmax. Intravenous administration of pertuzumab from GD19 through GD50 (period of organogenesis) was embryotoxic, with dose-dependent increases in embryo-foetal death between GD25 to GD70. The incidences of embryo-foetal loss were 33, 50, and 85% for pregnant female monkeys treated with bi weekly pertuzumab doses of 10, 30, and 100 mg/kg, respectively (4- to 35-fold greater than the recommended human dose, based on Cmax). At Caesarean section on GD100, oligohydramnios, decreased relative lung and kidney weights and microscopic evidence of renal hypoplasia consistent with delayed renal development were identified in all pertuzumab dose groups. In addition, consistent with foetal growth restrictions, secondary to oligohydramnios, lung hypoplasia (1 of 6 in 30 mg/kg and 1 of 2 in 100 mg/kg groups), ventricular septal defects (1 of 6 in 30 mg/kg group), thin ventricular wall (1 of 2 in 100 mg/kg group) and minor skeletal defects (external -3 of 6 in 30 mg/kg group) were also noted. Pertuzumab exposure was reported in offspring from all treated groups, at levels of 29% to 40% of maternal serum levels at GD100.
Subcutaneous pertuzumab (250 mg/kg/week for 4 weeks) and intravenous pertuzumab (up to 150 mg/kg weekly for up to 26 weeks) was well tolerated in cynomolgus monkeys (binding species), except for the development of diarrhoea. With intravenous pertuzumab doses of 15 mg/kg and higher, intermittent mild treatment-associated diarrhoea was noted. In a subset of monkeys, chronic dosing (26 weekly doses) resulted in episodes of severe secretory diarrhoea. The diarrhoea was managed (with the exception of euthanasia of one animal, 50 mg/kg/dose) with supportive care including intravenous fluid replacement therapy.
Trastuzumab
Reproduction studies have been conducted in Cynomolgus monkeys via the intravenous route at doses up to 16 times that of the human maintenance trastuzumab dose in Phesgo of 600 mg formulation and have revealed no evidence of impaired fertility or harm to the foetus. Placental transfer of trastuzumab during the early (days 20-50 of gestation) and late (days 120-150 of gestation) foetal development period was observed.
There was no evidence of acute or multiple dose-related toxicity in studies of up to 6 months, or reproductive toxicity in teratology, female fertility or late gestational toxicity/placental transfer studies. Trastuzumab is not genotoxic. A study of trehalose, a major formulation excipient did not reveal any toxicities.
No long-term animal studies have been performed to establish the carcinogenic potential of trastuzumab, or to determine its effects on fertility in males.
A study conducted in lactating Cynomolgus monkeys administered intravenous trastuzumab doses up to 16 times that of the human maintenance dose of 600 mg trastuzumab in the Phesgo formulation demonstrated that trastuzumab is secreted in the milk post partum. The exposure to trastuzumab in utero and the presence of trastuzumab in the serum of infant monkeys was not associated with any adverse effects on their growth or development from birth to 1 month of age.
Hyaluronidase
Hyaluronidase is found in most tissues of the human body. Non-clinical data for recombinant human hyaluronidase reveal no special hazard for humans based on conventional studies of repeated dose toxicity including safety pharmacology endpoints. Reproductive toxicology studies with vorhyaluronidase alfa revealed embryofetal toxicity in mice at high systemic exposure, but did not show teratogenic potential.
A single dose study in rabbits and a 13-week repeat dose toxicity study in Cynomolgus monkeys were conducted with trastuzumab subcutaneous formulation. The rabbit study was performed to specifically examine local tolerance aspects. The 13-week study was performed to confirm that the change to the subcutaneous route of administration and the use of the excipient vorhyaluronidase alfa did not have an effect on the trastuzumab safety characteristics. Trastuzumab subcutaneous formulation was locally and systemically well tolerated.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.