PIASKY Solution for injection/infusion Ref.[111488] Active ingredients: Crovalimab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, Complement inhibitors
ATC code: L04AJ07
Mechanism of action
Crovalimab is a recombinant humanised immunoglobulin G1 (IgG1)-based monoclonal antibody that specifically binds with high affinity to component 5 (C5) of the complement system, inhibiting its cleavage into C5a and C5b and thus preventing the formation of the membrane attack complex (MAC). Crovalimab causes terminal complement activity inhibition. In patients with PNH, crovalimab inhibits terminal complement-mediated intravascular haemolysis.
Pharmacodynamic effects
In clinical studies with PNH patients, a concentration-dependent inhibition of terminal complement activity following treatment with crovalimab was observed. Terminal complement activity (CH50 as measured by Liposome Immunoassay [LIA]) inhibition was achieved immediately by the end of the initial crovalimab infusion and was generally sustained through the duration of crovalimab treatment.
Similarly, mean free C5 concentrations decreased to low levels (<0.0001 g/L) in comparison to baseline and remained low throughout the treatment period.
Free C5 and CH50 levels were similar between paediatric and adult patients treated with crovalimab.
Clinical efficacy and safety
The safety and efficacy of crovalimab in patients with PNH were evaluated in a non-inferiority Phase III study (COMMODORE 2, BO42162) and supported by clinical evidence from two additional Phase III studies (COMMODORE 3, YO42311 and COMMODORE 1, BO42161).
In all Phase III studies, patients were required to be vaccinated against Neisseria meningitidis, either within 3 years prior to the start of treatment or within 7 days after starting treatment with crovalimab. Patients vaccinated within 2 weeks prior to initiating crovalimab or after the start of study treatment received appropriate prophylactic antibiotics from the time they started Piasky until at least 2 weeks after the vaccination (see section 4.4 for warnings and precautions related to serious meningococcal infection). Patients with a history of Neisseria meningitidis infection in the 6 months prior to screening and up to the first study drug administration were excluded.
Patients were also excluded if they had a history of allogenic bone marrow transplantation.
Crovalimab was administered in Phase III studies in accordance with the recommended dose described in section 4.2. Rescue doses of 340 mg of crovalimab administered intravenously were allowed based on the investigators' judgement if a patient experienced signs and symptoms of PNH; however, these studies were not designed to evaluate the impact of rescue dosing on the efficacy of crovalimab. Eculizumab was administered per local prescribing information, or in a country without access to commercial eculizumab (COMMODORE 2), then eculizumab 600 mg was given intravenously once weekly for the first 4 weeks, followed by 900 mg every 2 weeks thereafter. Rescue doses of eculizumab were not allowed on study.
The Phase III studies consisted of a primary treatment period of 24 weeks, after which patients had the option to continue/switch to crovalimab in an extension period.
Study in complement inhibitor-naïve patients with PNH
COMMODORE 2 (Study BO42162)
COMMODORE 2 was a Phase III, randomised, open-label, active-controlled, multicentre clinical study designed to evaluate the efficacy and safety of crovalimab compared to eculizumab in patients with PNH who were not previously treated with a complement inhibitor. 204 patients (body weight ≥40 kg), were randomised 2:1 to receive either crovalimab (n=135) or eculizumab (n=69). The study additionally enrolled 6 paediatric patients (aged <18 years and with body weight ≥40 kg) in a descriptive arm to receive crovalimab (see section 5.1). Eligible patients had high disease activity at screening, demonstrated by LDH level ≥2 × upper limit of normal (ULN) and by the presence of one or more PNH-related signs or symptoms in the past 3 months: fatigue, haemoglobinuria, abdominal pain, shortness of breath (dyspnoea) anaemia (haemoglobin <10 g/dL), history of a major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction; or history of packed red blood cell (pRBC) transfusion due to PNH.
Randomisation was stratified by the most recent LDH value (≥2 to ≤4 × ULN, or >4 × ULN) and by the transfusion history (0, >0 to ≤6, or >6 pRBC units administered within 6 months prior to randomisation); the respective stratification categories were balanced across treatment arms.
Demographics and baseline characteristics of the randomised study population were generally balanced between the treatment arms and are presented in Table 3.
Table 3. Demographics and baseline characteristics for COMMODORE 2 (randomised population):
| Parameters | Crovalimab (N=135) | Eculizumab (N=69) |
|---|---|---|
| Age (years) at PNH diagnosis | ||
| Mean (SD) Median (Range) | 35.8 (15.5) 31.0 (11.5-74.7) | 37.4 (16.4) 32.1 (11.2-76.8) |
| Age (years) at first administration of the study treatment* | ||
| Mean (SD) Median (Range) | 40.5 (15.2) 36.0 (18-76) | 41.9 (16.0) 38.0 (17-78) |
| <18 years (n, %) 18-64 years (n, %) ≥65 years (n, %) | 0 122 (90.4%) 13 (9.6%) | 2 (2.9%) 58 (84.1%) 9 (13.0%) |
| Weight | ||
| 40 <100 kg (n, %) ≥100 kg (n, %) | 131 (97.0%) 4 (3.0%) | 66 (95.7%) 3 (4.3%) |
| Sex | ||
| Male (n, %) Female (n, %) | 77 (57.0%) 58 (43.0%) | 35 (50.7%) 34 (49.3%) |
| LDH levels at baseline (x ULN) | ||
| Median (Range) | 7.0 (2.0-16.3) | 7.7 (2.0-20.3) |
| History of pRBC transfusions in the 12 months prior to screening | ||
| Yes (n, %) | 103 (77.4%) | 50 (73.5%) |
| pRBC units transfused in the 12 months prior to screening | ||
| Median (Range) | 3.8 (0-43.5) | 3.0 (0-41.0) |
| Total PNH granulocyte clone size (%) | ||
| Median (Range) | 91.4 (5.8-100) | 93.6 (6.8-99.9) |
| Total PNH monocyte clone size (%) | ||
| Median (Range) | 90.9 (42.5-99.9) | 95.1 (41.5-99.9) |
| Total PNH erythrocytes clone size (%) | ||
| Median (Range) | 25.3 (3.5-96.0) | 44.6 (0.1-88.9) |
| Haemoglobin levels at baseline (g/L) | ||
| Median (IQR) | 85.0 (77.0-93.0) | 87.0 (81.0-97.0) |
| History of aplastic anaemia | ||
| Yes (n, %) | 53 (39.3%) | 26 (37.7%) |
| History of myelodysplastic syndrome | ||
| Yes (n, %) | 6 (4.4%) | 6 (8.7%) |
| History of Major Adverse Vascular Event (MAVE) | ||
| Yes (n, %) | 21 (15.6%) | 10 (14.5%) |
| Medicinal products at baseline** | ||
| Anticoagulants (n, %) Steroids (n, %) Immunosuppressive therapy (n, %) | 35 (25.9%) 46 (34.1%) 23 (17.0%) | 17 (24.6%) 25 (36.2%) 13 (18.8%) |
| PNH-related signs or symptoms within 3 months prior to screening | ||
| Abdominal Pain | 21 (15.6%) | 11 (15.9%) |
| Anaemia | 109 (80.7%) | 57 (82.6%) |
| Dysphagia | 8 (5.9%) | 2 (2.9%) |
| Erectile Dysfunction | 13 (9.6%) | 4 (5.8%) |
| Fatigue | 113 (83.7%) | 63 (91.3%) |
| Haemoglobinuria | 79 (58.5%) | 45 (65.2%) |
| MAVE (including Thrombosis) | 9 (6.7%) | 5 (7.2%) |
| Shortness of Breath (Dyspnoea) | 29 (21.5%) | 14 (20.3%) |
Note: IQR = interquartile range.
* Two adolescent patients (both 17 years of age) were randomised into the eculizumab arm prior to the opening of the separate descriptive paediatric arm. Both patients switched to crovalimab in the extension period after completing the primary treatment period; one patient was still <18 years, while the other patient had turned 18 years at the time of first crovalimab treatment. See below "Paediatric population"
** Includes medicinal products that were started prior to initiation of study treatment, and were either stopped before or were ongoing at time of initiation of study treatment.
The primary objective of the study was to evaluate the efficacy of crovalimab compared with eculizumab, based on the non-inferiority (NI) assessment of the following co-primary endpoints: haemolysis control, measured by the mean proportion of patients with LDH ≤1.5x ULN from Week 5 to Week 25; and the proportion of patients who achieved transfusion avoidance, defined as patients who are pRBC transfusion-free, from baseline through Week 25. Secondary efficacy endpoints included the proportion of patients with breakthrough haemolysis, proportion of patients with stabilised haemoglobin, and change in fatigue (measured by the FACIT [Functional Assessment of Chronic Illness Therapy]-Fatigue scale) from baseline to Week 25.
Crovalimab was non-inferior compared to eculizumab for both co-primary endpoints of haemolysis control and transfusion avoidance and for the secondary endpoints of haemoglobin stabilisation and breakthrough haemolysis (Figure 1). Figure 2 shows the proportion of patients with LDH ≤1.5 ×ULN from baseline through Week 25.
Figure 1. Co-primary and secondary endpoint results in (COMMODORE 2, primary analysis population):
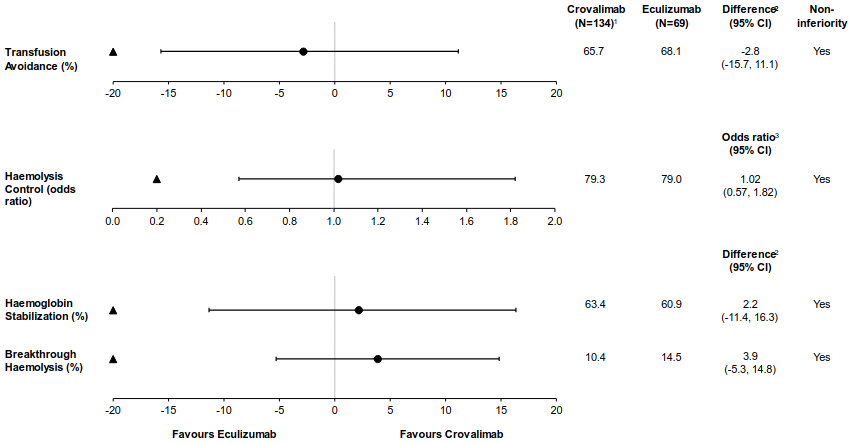
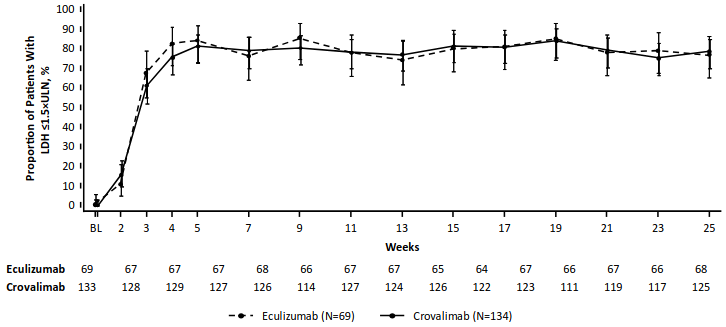
Figure 2. Proportion of patients with LDH ≤1.5 × ULN from baseline through Week 25, with 95% CIs (COMMODORE 2, primary analysis population):
Studies in PNH patients previously treated with complement C5 inhibitor therapy
COMMODORE 1 (Study BO42161) - randomised eculizumab switch patients
COMMODORE 1 was a Phase III, randomised, open-label, active-controlled, multicentre clinical study evaluating the safety, pharmacodynamics, pharmacokinetics and exploratory efficacy of crovalimab in patients switching from another complement C5 inhibitor therapy. The primary objective of this study was to evaluate safety (see section 4.8). 89 patients were randomised 1:1 to receive either crovalimab (n=45) or eculizumab (n=44). Patients were eligible to enroll into the randomised arms if they were switching from approved doses of eculizumab and had haemolysis control at screening, defined by LDH level ≤1.5 × ULN. Patients were excluded if they had a Major Adverse Vascular Event (MAVE) within the 6 months prior to first study drug administration. Randomisation was stratified by patient transfusion history (whether a patient received a transfusion of pRBCs within 12 months prior to randomisation).
Demographics and baseline characteristics of the randomised study population were balanced between the treatment arms. The median LDH value at baseline was 1.01 x ULN (range: 0.6-1.7) for crovalimab and 0.96 × ULN (range: 0.7-1.9) for eculizumab. The proportion of patients with a history of transfusions in the 12 months prior to screening was 22.7% in the crovalimab arm and 25% in the eculizumab arm, with a mean (SD) of 1.6 (3.7) and 2.3 (5.4) units of transfused pRBC in the crovalimab and eculizumab arms respectively. The baseline median (range) PNH clone sizes for total erythrocytes, monocytes, and granulocytes for crovalimab arm vs eculizumab arms are as follows: 44.6% (2.6-100) vs 54.2% (1.3-100), 88.6% (13.8-100) vs 96.4% (7.6-99.9), and 88.1% (5.2-100), vs 95.7% (7.9-99.9), respectively.
Out of 89 randomised patients, efficacy was evaluated in an exploratory fashion in 76 (n=39 for crovalimab and n=37 for eculizumab) that were enrolled at least 24 weeks before the cut-off date for the primary analysis. Overall, the results of the exploratory efficacy endpoints showed that patients switching to crovalimab from eculizumab maintained disease control. The mean proportion of patients maintaining haemolysis control from baseline through Week 25 was 92.9% [95% CI: 86.6, 96.4] for patients randomised to crovalimab and 93.7% [95% CI: 87.3, 97.0] for patients randomised to eculizumab. Transfusion avoidance was observed in 79.5% [95% CI: 63.1, 90.1] of patients randomised to crovalimab and 78.4% [95% CI: 61.3, 89.6] of patients randomised to eculizumab.
COMMODORE 1 (Study BO42161) and COMMODORE 2 (Study BO42162) - clinically stable switch patients
Supportive data in clinically stable eculizumab switch patients was reported from patients in COMMODORE 1 (25 efficacy evaluable patients) and COMMODORE 2 (29 efficacy evaluable patients) that had been treated with eculizumab for at least 24 weeks in the primary treatment period and had LDH ≤1.5 × ULN at switch-to-crovalimab baseline.
Efficacy was evaluated in the patients that had at least 24 weeks of exposure to crovalimab (or otherwise discontinued prior to having reached 24 weeks of treatment). The mean proportion of clinically stable switch patients maintaining haemolysis control from switch baseline through switch Week 25 in COMMODORE 1 and COMMODORE 2 was 98.7% [95% CI: 96.2, 99.5] and 95.3% [95% CI: 89.5, 97.9], respectively. Transfusion avoidance was observed in 80.0% [95% CI: 58.70, 92.39] and 86.2% [95% CI: 67.43, 95.49] of the clinically stable switch patients, respectively. These results in clinically stable eculizumab switch patients were consistent with the results in randomised eculizumab switch patients during the primary treatment period of COMMODORE 1.
Furthermore, in the non-randomised arm of COMMODORE 1, of the 19 clinically stable patients switching from ravulizumab, 95.8% [95%CI: 89.11, 98.43] maintained haemolysis control and 57.9% [95% CI: 33.97, 78.88] patients were transfusion avoidant from baseline to Week 25.
Immunogenicity
As with all therapeutic proteins, there is the potential for immune response to crovalimab.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medicinal products and underlying disease. For these reasons, comparison of incidence of antibodies to crovalimab with the incidence of antibodies to other products may be misleading.
In the Phase III study COMMODORE 2, treatment-emergent anti-drug antibodies (ADAs) were observed in 35.0% (49/140) of treatment-naïve patients who received crovalimab and 38.2% (26/68) of patients who switched from treatment with another C5 inhibitor to crovalimab. The median time to the development of first post-baseline ADAs was 16.1 weeks (range: 1.1 to 72.3 weeks), and 16.6 weeks (range: 2.1 to 36.3 weeks) in the treatment-naïve patients and patients who were previously treated with another C5 inhibitor, respectively. Across Phase III studies, the incidence of treatment-emergent ADAs was 35.1% (67 patients out of 191) and 25.4% (51 patients out of 201) in treatment-naive and patients who switched from treatment with another C5 inhibitor to crovalimab, respectively.
Across Phase III studies, median concentration time-courses in ADA-positive patients were slightly lower in comparison to ADA-negative patients. Despite this effect, concentrations remained above 100 μg/mL (threshold for complete terminal complement inhibition) in more than 80% of ADA-positive patients. ADA presence was not associated with clinically meaningful impact on pharmacokinetics, pharmacodynamics, and efficacy in most of the patients. However, out of 392 patients evaluated for ADA status, partial or complete loss of exposure associated with ADA onset was observed in 23 patients (5.9%); among them, 17 (4.3%) ADA positive patients had a loss of pharmacological activity (based on CH50 or free C5) coinciding with a loss of exposure, with loss of efficacy, manifesting as a sustained loss of haemolysis control in 7 patients (1.8%). There was no evidence for a clinical impact of ADA status on the safety profile of Piasky (see sections 4.4 and 4.8).
Paediatric population
Ten paediatric patients (with body weight ≥40 kg) treated with crovalimab in COMMODORE 2 (n=7; 13-17 years old) and COMMODORE 3 (n=3; 15-17 years old) were evaluable for efficacy. Nine patients were treatment-naive and 1 patient switched from eculizumab to crovalimab in the extension period. All paediatric patients received the same dosing as adult patients based on body weight. All 9 treatment-naive patients achieved haemolysis control (defined as LDH ≤1.5 x ULN) by Week 4 and this was maintained in 7 patients at each visit from baseline to Week 25; the patient switching from eculizumab to crovalimab maintained haemolysis control through 24 weeks of treatment in the extension period. Seven out of the 10 paediatric patients achieved transfusion avoidance and haemoglobin stabilisation, and no patients had a breakthrough haemolysis event during the 24-week treatment period.
Overall, the treatment effect of crovalimab in paediatric PNH patients was similar to that observed in adult PNH patients.
The European Medicines Agency has deferred the obligation to submit the results of studies with Piasky in one or more subsets of the paediatric population with PNH (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of crovalimab have been characterised both in healthy volunteers and in patients with PNH. The pharmacokinetics were characterised using non-linear mixed effects pharmacokinetic analysis methods, based on a pooled database composed of 9 healthy volunteers and 210 and 211 treatment-naïve patients and patients who switched from previous treatment with another C5 inhibitor to crovalimab, respectively.
The concentration-time course of crovalimab is best described using a two-compartment open model with first-order elimination and a first order subcutaneous absorption constant. To describe the transient increase in clearance due to the formation of Type III immune complexes observed in patients who switched from treatment with another C5 inhibitor to crovalimab, an additional time-varying clearance parameter, which decreases exponentially with time, was added. At steady state, exposure is expected to be similar between treatment naïve and switch patients.
Absorption
The absorption rate constant was estimated to be 0.126 day -1 [CV%: 38.3]. Following subcutaneous administration, the bioavailability was estimated at 83.0% [CV%: 116].
Distribution
The central volume of distribution was estimated to be 3.23 L [CV%: 22.4] and the peripheral volume of distribution was estimated as 2.32 L [CV%: 70.6].
The small volume of distribution indicates that crovalimab is likely to be distributed mainly in serum and/or in vascular rich tissues.
Biotransformation
The metabolism of crovalimab has not been directly studied. IgG antibodies are mainly catabolised by lysosomal proteolysis and then eliminated from or reused by the body.
Elimination
The clearance was estimated to be 0.0791 L/day [CV%: 20.6]. The terminal half-life of crovalimab was estimated as 53.1 days [CV%: 39.9], which is longer compared to other humanised IgG antibodies. This long half-life is consistent with the recycling properties of crovalimab.
Special populations
No pharmacokinetic studies with crovalimab have been conducted in special populations. Bodyweight was shown to be a significant covariate, with clearances and volumes of distribution increasing and crovalimab exposure decreasing as bodyweight increases. Therefore, posology of crovalimab is based on the bodyweight of the patient (see section 4.2).
After inclusion of bodyweight in the model, the population pharmacokinetics analyses in patients with PNH showed that age (13–85 years) and gender did not meaningfully influence the pharmacokinetics of crovalimab. No further dose adjustment is required.
Race/ethnicity was also shown not to have an impact on the pharmacokinetics of crovalimab; however, data are limited in Black patients and therefore not considered conclusive in this population.
Elderly
No dedicated studies have been conducted to investigate the pharmacokinetics of crovalimab in patients aged ≥65 years, however 46 (10.9%) elderly PNH patients were enrolled in clinical studies, including 35 patients aged 65-74 years, 10 patients aged 75-84 years, and 1 patient aged ≥85 years. The data obtained in PNH clinical studies indicates that exposure in patients aged ≥65 years is comparable to that of younger patients in other age groups, however, due to the limited data in patients ≥85 years, the pharmacokinetics of crovalimab in those subjects is unknown.
Renal impairment
No dedicated studies have been conducted to investigate the pharmacokinetics of crovalimab in patients with renal impairment, however the data obtained in PNH clinical studies (62 [14.7%] patients with mild renal impairment, 38 [9%] patients with moderate renal impairment, and 4 [1%] patients with severe renal impairment) indicate that exposure in patients with mild, moderate, or severe renal impairment is comparable to that of patients without renal impairment. However, limited data were obtained for patients with severe renal impairment in PNH clinical studies.
Hepatic impairment
No dedicated studies have been conducted in patients with hepatic impairment, however data obtained in PNH clinical studies indicate that exposure in patients with mild hepatic impairment (46 [11%] as graded based on alanine aminotransferase levels) are comparable to that of patients without hepatic impairment. Limited pharmacokinetic data were available in PNH patients with moderate (0 [0%]) to severe (1[0.23%]) hepatic impairment, therefore the impact of moderate or severe hepatic impairment on the pharmacokinetics of crovalimab is unknown and no dose recommendation can be provided (see section 4.2).
Paediatric population
Data obtained in 12 paediatric patients (13-17 years old) in the PNH clinical studies indicates that exposure in paediatric patients 12 years of age or older with a weight of 40 kg and above was found to be comparable to that of adult patients.
5.3. Preclinical safety data
Non-clinical data revealed no special hazard related to crovalimab treatment for humans based on conventional studies of, repeated dose toxicity (including safety pharmacology endpoints), and toxicity to reproduction and development.
Genotoxicity
No dedicated studies have been performed to establish the genotoxic potential of crovalimab. Monoclonal antibodies are not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity
No studies have been performed to establish the carcinogenic potential of crovalimab. Assessment of available evidence related to pharmacodynamic effects and animal toxicology data do not indicate carcinogenic potential of crovalimab.
Reproductive and developmental toxicity
Repeated administration of crovalimab to pregnant cynomolgus monkeys during the gestation period induced no maternal toxicity and did not affect pregnancy outcome. No effects on the viability, growth and development of the infants were observed during the 6-month postnatal period.
Fertility
No effects on female or male reproductive organs were observed in cynomolgus monkeys following repeated administration of crovalimab for up to 6 months. Separate animal fertility studies have not been conducted with crovalimab.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.