PLUVICTO Solution for injection/infusion Ref.[50608] Active ingredients: Lutetium ¹⁷⁷Lu vipivotide tetraxetan
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Therapeutic radiopharmaceuticals, Other therapeutic radiopharmaceuticals
ATC code: V10XX05
Mechanism of action
The active moiety of Pluvicto is the radionuclide lutetium-177 which is linked to a small-molecule ligand that targets and binds with high affinity to PSMA, a transmembrane protein that is highly expressed in prostate cancer, including mCRPC. Upon the binding of Pluvicto to PSMA-expressing cancer cells, the beta-minus emission from lutetium-177 delivers therapeutic radiation to the targeted cell, as well as to surrounding cells, and induces DNA damage which can lead to cell death.
Pharmacodynamic effects
Unlabelled vipivotide tetraxetan does not have any pharmacodynamic activity.
Clinical efficacy and safety
VISION
The efficacy of Pluvicto in patients with progressive, PSMA-positive mCRPC was evaluated in VISION, a randomised, multicentre, open-label phase III study. Eight hundred and thirty-one (N=831) adult patients were randomised (2:1) to receive either Pluvicto 7 400 MBq every 6 weeks for up to a total of 6 doses plus best standard of care (BSoC) (N=551) or BSoC alone (N=280). Patients who received 4 doses of Pluvicto were reassessed for evidence of response, signs of residual disease, and tolerability and could receive up to 2 additional doses per physician’s discretion.
To maintain castration status, all patients continued to receive a GnRH analogue or had prior bilateral orchiectomy. Eligible patients were required to have progressive, PSMA-positive mCRPC, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 to 2, at least one metastatic lesion present on computed tomography (CT), magnetic resonance imaging (MRI) or bone scan imaging, and adequate renal, hepatic and haematological function.
Eligible patients were also required to have received at least one AR pathway inhibitor, such as abiraterone acetate or enzalutamide, and 1 or 2 prior taxane-based chemotherapy regimens (with a regimen defined as a minimum exposure of 2 cycles of a taxane). Patients treated with only 1 prior taxane-based chemotherapy regimen were eligible if the patient was unwilling or the physician deemed the patient unsuitable to receive a second regimen. Patients with unstable symptomatic central nervous system metastases or symptomatic or clinically/radiologically impending spinal cord compression were not eligible for the study. Patients underwent a gallium (68Ga) gozetotide positron emission tomography (PET) scan to evaluate PSMA expression in lesions defined by central read criteria. Eligible patients were required to have PSMA-positive mCRPC defined as having at least one tumour lesion with gallium (68Ga) gozetotide uptake greater than in normal liver. Patients were excluded if any lesions exceeding size criteria in short axis (organs ≥1 cm, lymph nodes ≥2.5 cm, bones [soft-tissue component] ≥1 cm) had uptake less than or equal to uptake in normal liver.
BSoC administered at the physician’s discretion included: supportive measures including pain management, hydration, blood transfusions, etc.; ketoconazole; radiation therapy (including seeded form or any external beam radiotherapy [including stereotactic body radiotherapy and palliative external beam]) to localised prostate cancer targets; bone-targeted agents including zoledronic acid, denosumab and any bisphosphonates; androgen-reducing agents including GnRH analogues, any corticosteroid, and 5-alpha reductases; AR pathway inhibitors. BSoC excluded investigational agents, cytotoxic chemotherapy, immunotherapy, other systemic radioisotopes and hemi-body radiotherapy treatment.
Patients continued randomised treatment until evidence of tumour progression (based on investigator assessment per Prostate Cancer Working Group 3 [PCWG3] criteria), unacceptable toxicity, use of prohibited treatment, non-compliance or withdrawal, or lack of clinical benefit.
The primary efficacy endpoints were overall survival (OS) and radiographic progression-free survival (rPFS) as determined by blinded independent central review (BICR) per PCWG3 criteria. Among the secondary efficacy endpoints were overall response rate (ORR) as determined by BICR per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 and time to first symptomatic skeletal event (SSE) defined as first new symptomatic pathological bone fracture, spinal cord compression, tumour-related orthopaedic surgical intervention, requirement for radiation therapy to relieve bone pain, or death from any cause, whichever occurred first. Radiographic imaging for tumour assessment (CT with contrast/MRI imaging and bone scan) was done every 8 weeks (±4 days) after the first dose for the first 24 weeks (independent of dose delays), then every 12 weeks (±4 days).
Demographic and baseline disease characteristics were balanced between the treatment arms. The median age was 71 years (range: 40 to 94 years); 86.8% White; 6.6% Black or African American; 2.4% Asian; 92.4% had ECOG PS0-1; 7.6% had ECOG PS2. Randomisation was stratified by baseline lactate dehydrogenase (LDH ≤260 IU/L vs. >260 IU/L), presence of liver metastases (yes vs. no), ECOG PS score (0 or 1 vs. 2), and inclusion of an AR pathway inhibitor as part of BSoC at the time of randomisation (yes vs. no). At randomisation, all patients (100.0%) had received at least one prior taxane-based chemotherapy regimen and 41.2% of patients had received two; 97.1% of patients had received docetaxel and 38.0% of patients had received cabazitaxel. At randomisation, 51.3% of patients had received one prior AR pathway inhibitor, 41.0% of patients had received 2, and 7.7% of patients had received 3 or more. During the randomised treatment period, 52.6% of patients in the Pluvicto plus BSoC arm and 67.8% of patients in the BSoC alone arm received at least one AR pathway inhibitor.
Efficacy results for VISION are presented in Table 3 and Figures 1 and 2. The final analyses of OS and rPFS were event-driven and conducted after the occurrence of 530 deaths and 347 events, respectively.
Table 3. Efficacy results in VISION:
| Efficacy parameters | Pluvicto plus BsoC | BSoC |
|---|---|---|
| Alternate primary efficacy endpoints | ||
| Overall survival (OS)a | N=551 | N=280 |
| Deaths, n (%) | 343 (62.3%) | 187 (66.8%) |
| Median, months (95% CI)b | 15.3 (14.2; 16.9) | 11.3 (9.8; 13.5) |
| Hazard ratio (95% CI)c | 0.62 (0.52; 0.74) | |
| P-valued | <0.001 | |
| Radiographic progression-free survival (rPFS)e,f | N=385 | N=196 |
| Events (progression or death), n (%) | 254 (66.0%) | 93 (47.4%) |
| Radiographic progressions, n (%) | 171 (44.4%) | 59 (30.1%) |
| Deaths, n (%) | 83 (21.6%) | 34 (17.3%) |
| Median, months (99.2% CI)b | 8.7 (7.9; 10.8) | 3.4 (2.4; 4.0) |
| Hazard ratio (99.2% CI)c | 0.40 (0.29; 0.57) | |
| P-valued | <0.001 | |
| Secondary efficacy endpoints | ||
| Time to first symptomatic skeletal event (SSE)f | N=385 | N=196 |
| Events (SSE or death), n (%) | 256 (66.5%) | 137 (69.9%) |
| SSEs, n (%) | 60 (15.6%) | 34 (17.3%) |
| Deaths, n (%) | 196 (50.9%) | 103 (52.6%) |
| Median, months (95% CI)b | 11.5 (10.3; 13.2) | 6.8 (5.2; 8.5) |
| Hazard ratio (95% CI)c | 0.50 (0.40; 0.62) | |
| P-valueg | <0.001 | |
| Best overall response (BOR) | ||
| Patients with evaluable disease at baseline | N=319 | N=120 |
| Complete response (CR), n (%) | 18 (5.6%) | 0 (0%) |
| Partial response (PR), n (%) | 77 (24.1%) | 2 (1.7%) |
| Overall response rate (ORR)h,i | 95 (29.8%) | 2 (1.7%) |
| P-valuej | <0.001 | |
| Duration of response (DOR)h | ||
| Median, months (95% CI)b | 9.8 (9.1; 11.7) | 10.6 (NE; NE)k |
BSoC: Best standard of care; CI: Confidence interval; NE: Not evaluable; BICR: Blinded independent central review; PCWG3: Prostate Cancer Working Group 3; RECIST: Response Evaluation Criteria in Solid Tumors.
a Analysed on an intent-to-treat (ITT) basis in all randomised patients.
b Based on Kaplan-Meier estimate.
c Hazard ratio based on the stratified Cox PH model. Hazard ratio <1 favours Pluvicto plus BSoC.
d Stratified log-rank test one-sided p-value.
e By BICR per PCWG3 criteria. The primary analysis of rPFS included censoring of patients who had ≥2 consecutive missed tumour assessments immediately prior to progression or death. Results for rPFS with and without censoring for missed assessments were consistent.
f Analysed on an ITT basis in all patients randomised on or after 05-Mar-2019, when actions were implemented to mitigate early drop-out from BSoC arm.
g Stratified log-rank test two-sided p-value.
h By BICR per RECIST v1.1.
i ORR: CR+PR. Confirmed response for CR and PR.
j Stratified Wald’s Chi-square test two-sided p-value.
k Median DOR in the BSoC only arm was not reliable since only 1 of the 2 patients who responded had RECIST v1.1 radiographic progression or death.
Figure 1. Kaplan-Meier plot of OS in VISION:
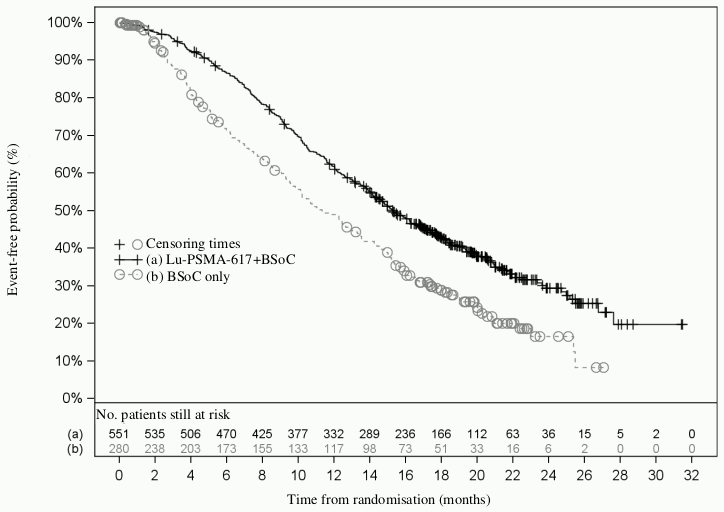
Stratified log-rank test and stratified Cox model using strata per Interactive Response Technology (IRT) defined by LDH level, presence of liver metastases, ECOG score and inclusion of an AR pathway inhibitor in BSoC at time of randomisation.
n/N: Number of events/number of patients in treatment arm.
Figure 2. Kaplan-Meier plot of BICR-assessed rPFS in VISION:
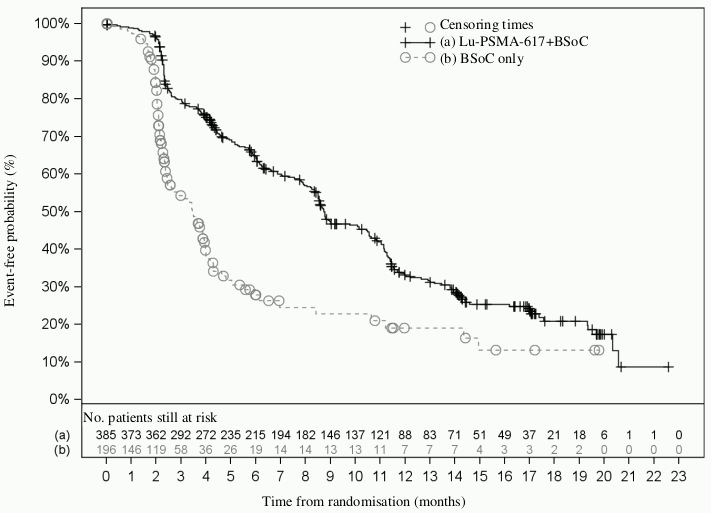
Stratified log-rank test and stratified Cox model using strata per IRT defined by LDH level, presence of liver metastases, ECOG score and inclusion of an AR pathway inhibitor in BSoC at time of randomisation.
n/N: Number of events/number of patients in treatment arm.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Pluvicto in all subsets of the paediatric population in the treatment of PSMA-expressing prostate cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of lutetium (177Lu) vipivotide tetraxetan have been characterised in 30 patients in the phase III VISION sub-study.
Absorption
Pluvicto is administered intravenously and is immediately and completely bioavailable.
The geometric mean blood exposure (area under the curve [AUCinf]) for lutetium (177Lu) vipivotide tetraxetan at the recommended dose is 52.3 ng.h/mL (geometric mean coefficient of variation [CV] 31.4%). The geometric mean maximum blood concentration (Cmax) for lutetium (177Lu) vipivotide tetraxetan is 6.58 ng/mL (CV 43.5%).
Distribution
The geometric mean volume of distribution (Vz) for lutetium (177Lu) vipivotide tetraxetan is 123 L (CV 78.1%).
Unlabelled vipivotide tetraxetan and non-radioactive lutetium (175Lu) vipivotide tetraxetan are each 60% to 70% bound to human plasma proteins.
Organ uptake
The biodistribution of lutetium (177Lu) vipivotide tetraxetan shows primary uptake in lacrimal glands, salivary glands, kidneys, urinary bladder wall, liver, small intestine and large intestine (left and right colon).
Elimination
The geometric mean clearance (CL) for lutetium (177Lu) vipivotide tetraxetan is 2.04 L/h (CV 31.5%).
Lutetium (177Lu) vipivotide tetraxetan is primarily eliminated renally.
Half-life
Pluvicto shows a bi-exponential elimination with a geometric mean terminal elimination half-life (t½) of 41.6 hours (CV 68.8%).
Biotransformation
Lutetium (177Lu) vipivotide tetraxetan does not undergo hepatic or renal metabolism.
In vitro evaluation of drug interaction potential
CYP450 enzymes
Vipivotide tetraxetan is not a substrate of cytochrome P450 (CYP450) enzymes. It does not induce cytochrome P450 (CYP) 1A2, 2B6 or 3A4, and it does not inhibit cytochrome P450 (CYP) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 3A4/5 in vitro.
Transporters
Vipivotide tetraxetan is not a substrate of BCRP, P-gp, MATE1, MATE2-K, OAT1, OAT3 or OCT2, and it is not an inhibitor of BCRP, P-gp, BSEP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 or OCT2 in vitro.
Special populations
Effects of age and body weight
No clinically significant effects on the pharmacokinetic parameters of lutetium (177Lu) vipivotide tetraxetan were identified for the following covariates assessed in 30 patients in the phase III VISION sub-study: age (median: 67 years; range: 52 to 80 years) and body weight (median: 88.8 kg; range: 63.8 to 143.0 kg).
Renal impairment
Exposure (AUC) of lutetium (177Lu) vipivotide tetraxetan increased by 20% in patients with mild renal impairment compared to normal renal function. Kidney dosimetry half-life also increased in patients with mild renal impairment compared to normal renal function, 51 hours vs. 37 hours, respectively. Patients with mild or moderate renal impairment may be at greater risk of toxicity (see section 4.4). No pharmacokinetic data are available for patients with moderate to severe renal impairment with baseline CLcr <50 mL/min or end-stage renal disease.
5.3. Preclinical safety data
No toxicological effects were observed in safety pharmacology or single-dose toxicity studies in rats and minipigs administered a non-radioactive formulation containing unlabelled vipivotide tetraxetan and lutetium (175Lu) vipivotide tetraxetan, or in repeat-dose toxicity studies in rats administered unlabelled vipivotide tetraxetan.
Carcinogenicity and mutagenicity
Mutagenicity and long-term carcinogenicity studies have not been carried out with lutetium (177Lu) vipivotide tetraxetan; however, radiation is a carcinogen and mutagen.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.