QINLOCK Tablet Ref.[28085] Active ingredients: Ripretinib
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Deciphera Pharmaceuticals (Netherlands) B.V., Atrium Building Floor 4th, Strawinskylaan 3051, 1077ZX, Amsterdam, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, other protein kinase inhibitors
ATC Code: L01EX19
Mechanism of action
Ripretinib is a novel tyrosine kinase inhibitor that inhibits KIT proto-oncogene receptor tyrosine kinase and PDGFRA kinase, including wild type, primary, and secondary mutations. Ripretinib also inhibits other kinases in vitro, such as PDGFRB, TIE2, VEGFR2, and BRAF.
Clinical efficacy and safety
INVICTUS (DCC-2618-03-001 study)
The efficacy and safety of QINLOCK were evaluated in a randomised (2:1), double-blind, placebo-controlled study (INVICTUS study) in patients with unresectable, locally advanced or metastatic GIST who had been previously treated with or are intolerant to at least 3 prior anticancer therapies including treatment with imatinib, sunitinib, and regorafenib. Randomisation was stratified by prior lines of therapy (3 versus ≥4) and Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1 or 2).
The primary efficacy outcome measure was progression-free survival (PFS) based on disease assessment by blinded independent central review (BICR) using modified RECIST 1.1 criteria, in which lymph nodes and bone lesions were not target lesions and a progressively growing new tumour nodule within a pre-existing tumour mass must meet specific criteria to be considered unequivocal evidence of progression. Secondary efficacy endpoints included objective response rate (ORR) by BICR, overall survival (OS), and patient-reported health state, physical function (PF), and role function (RF).
Participants were randomised to receive 150 mg QINLOCK (n=85) or placebo (n=44) orally once daily administered in continuous 28-day cycles. Treatment continued until disease progression or unacceptable toxicity. Individual treatment arms were unblinded at the time of disease progression as assessed by BICR review and all patients on placebo arm were offered to cross-over to QINLOCK. T he demographic characteristics were median age of 60 years (29 to 83 years) with 79 (61.2%) of patients aged 18-64 years, 32 (24.8%) of patients aged 65-74 years, and 18 (13.9%) patients aged ≥75 years (no patients ≥85 years old were randomised); male (56.6%); white (75.2%); and ECOG performance status of 0 (41.9%), 1 (49.6%), or 2 (8.5%). Sixty-three percent (63%) of patients received 3 prior therapies and approximately 37% received 4 or more prior therapies. Sixty-six percent (66%) of patients randomised to placebo crossed over to QINLOCK during the open-label period.
At the primary analysis (data cut-off date 31 May 2019) QINLOCK was compared to placebo in the INVICTUS study. QINLOCK demonstrated benefit in all assessed patient subgroups for PFS. Median PFS as determined by BICR (months) (95% CI) was 6.3 (4.6, 6.9) for QINLOCK versus 1.0 (0.9, 1.7) for placebo, HR (95% CI) 0.15 (0.09, 0.25) p-value <0.0001. The secondary endpoint ORR () was 9.4 (4.2, 18) for QINLOCK versus 0 (0, 8) for placebo, p-value 0.0504 and not statistically significant. Median OS (months) (95 CI) was 15.1 (12.3, 15.1) for QINLOCK versus 6.6 (4.1, 11.6) for placebo, nominal p-value 0.0004. OS was not evaluated for statistical significance as a result of the sequential testing procedure for the secondary endpoints of ORR and OS.
PFS, ORR and OS results from a more recent data cut-off (10 August 2020) are shown in Table 3 and Figures 1 and 2. PFS results were similar across subgroups based on age, sex, region, ECOG status and number of previous lines of therapy.
Table 3. INVICTUS efficacy results (as of 10 August 2020):
| QINLOCK (n=85) | Placebo (n=44) | |
|---|---|---|
| PFSa | ||
| Number of events (%) | 68 (80) | 37 (84) |
| Progressive disease | 62 (73) | 32 (73) |
| Deaths | 6 (7) | 5 (11) |
| Median PFS (months) (95% CI) | 6.3 (4.6, 8.1) | 1.0 (0.9, 1.7) |
| HR (95% CI)b | 0.16 (0.10, 0.27) | |
| ORRa | ||
| ORR (%) | 11.8 | 0 |
| (95% CI) | (5.8, 20.6) | (0, 8) |
| OS | ||
| Number of deaths (%) | 44 (52) | 35 (80) |
| Median OS (months) (95% CI) | 18.2 (13.1, NE) | 6.3 (4.1, 10.0) |
| HR (95% CI)b | 0.42 (0.27, 0.67) | |
BICR = Blinded Independent Central Review; CI = Confidence Interval; HR = Hazard Ratio; ORR = Objective Response Rate; NE = not estimable; PFS = Progression Free Survival; OS = Overall Survival
a Assessed per BICR.
b Hazard ratio is based on Cox proportional regression model. This model includes treatment and randomisation stratification factors as fixed factors.
Figure 1. INVICTUS Kaplan-Meier curve of progression-free survivala:
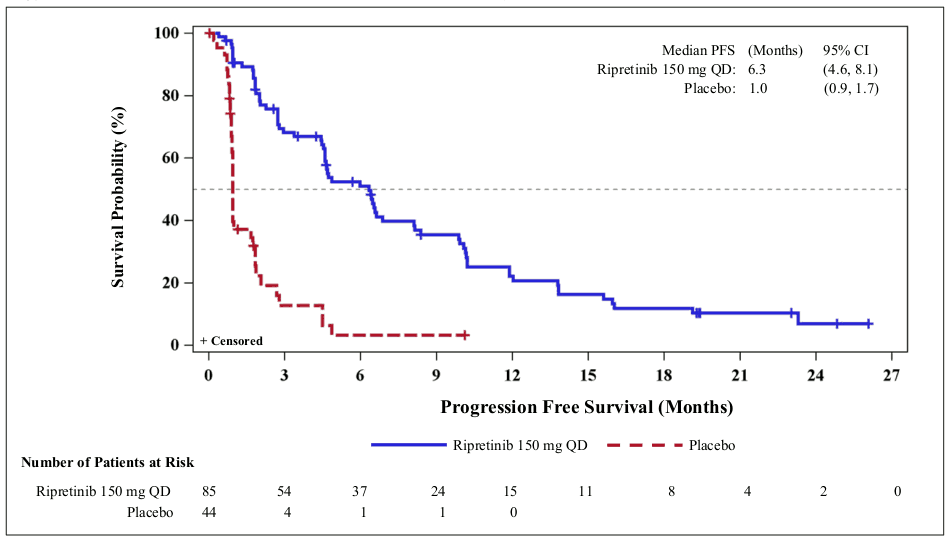
a Data cut off 10 August 2020
Figure 2. INVICTUS Kaplan-Meier curve of overall survivala:
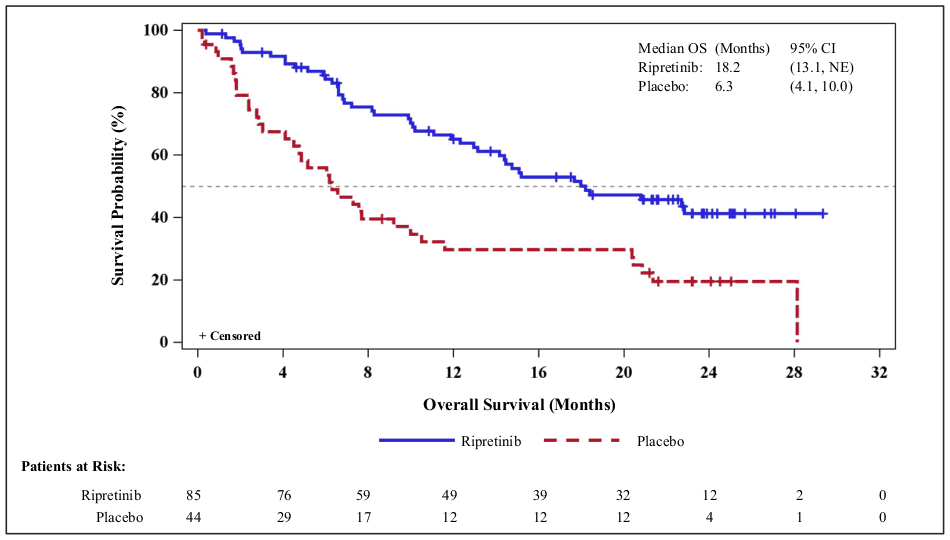
a Data cut off 10 August 2020
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with QINLOCK in all subsets of the paediatric population in the treatment of GIST (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Ripretinib reaches peak plasma concentrations at a median of 4 hours after oral administration of single dose ripretinib 150 mg (given as three tablets each containing 50 mg). The mean (CV%) AUC0-∞ after a single dose of 150 mg of ripretinib was 9,856 (39%) and 8,146 (56%) ng•h/mL for ripretinib and DP-5439, respectively.
Administration with a high-fat meal increased ripretinib AUC0-24 and Cmax by 30% and 22%, respectively. DP-5439 AUC0-24 and Cmax were higher by 47% and 66%, respectively.
Distribution
Both ripretinib and its active metabolite DP-5439 bind to plasma proteins at ≥99%. The mean (CV%) apparent volume of distribution (Vss/F) is approximately 302 (35%) L for ripretinib and 491 (38%) L for DP-5439.
Biotransformation
CYP3A4/5 is the major metaboliser of ripretinib and its active metabolite DP-5439, while CYP2C8 and CYP2D6 are minor metabolisers.
Elimination
Following oral administration of single dose ripretinib 150 mg in humans, mean (CV%) apparent oral clearance (CL/F) was 15.2 (39%) and 17.9 (56%) L/hr for ripretinib and DP-5439, respectively. Mean (CV%) half-life (t½) was 12.6 (17%) and 15.6 (23%) hours for ripretinib and DP-5439, respectively.
Systemic elimination of ripretinib was not primarily attributed to the kidney with 0.02% and 0.1% of the ripretinib dose excreted as ripretinib and DP-5439, respectively, in urine and 34% and 6% of the ripretinib dose excreted as ripretinib and DP-5439, respectively, in faeces.
Dose proportionality
Across the dose range of 20-250 mg, ripretinib and DP-5439 PK appeared to be less than dose proportional, especially at ripretinib doses higher than 150 mg.
Time dependency
Steady-state conditions are achieved within 14 days.
Specific populations
No clinically significant differences in the pharmacokinetics of QINLOCK were observed based on age (19 to 87 years), sex, race (White, Black, and Asian), body weight (39 to 138 kg), and tumour (GIST or other solid tumours).
Patients with renal impairment
In clinical studies, no relevant differences in exposure were observed between patients with mild and moderate renal impairment (CLcr 30 to 89 mL/min estimated by Cockcroft-Gault) and patients with normal renal function. Based on a population pharmacokinetic analysis, no dose adjustment is recommended in patients with mild and moderate renal impairment. The pharmacokinetics and safety of QINLOCK in patients with severe renal impairment (CLcr 15 to 29 mL/min estimated by Cockcroft-Gault) is limited. No dosing recommendation can be made in patients with severe renal impairment (see section 4.2).
Patients with hepatic impairment
In clinical studies, no relevant differences in exposure were observed between patients with mild (total bilirubin ≤ upper limit of normal (ULN) and AST > ULN, or total bilirubin > ULN to ≤1.5 × ULN and any AST) hepatic impairment and normal hepatic function. Based on a population pharmacokinetic analysis, no dose adjustment is recommended in patients with mild hepatic impairment. The pharmacokinetics and safety of QINLOCK in patients with moderate or severe hepatic impairment have not been studied; no dosing recommendation can be made in this subgroup (see section 4.2).
5.3. Preclinical safety data
The preclinical safety profile of ripretinib was assessed in rats and dogs for up to 13 weeks duration. Inflammation responses correlated with skin changes (discoloured, lesions) were recorded in rats (approximately 1.12 times the human exposure at 150 mg once daily). Elevated hepatic enzyme activity was reported in both species (approximately 1.12 and 1.3 times the human exposure at 150 mg once daily for rats and dogs, respectively). Dogs presented gastrointestinal effects (emesis and/or abnormal faeces) (approximately 1.3 times the human exposure at 150 mg once daily), and inflammatory responses illustrated by adverse skin lesions (approximately 0.14 times the human exposure at 150 mg once daily).
Carcinogenicity
Carcinogenicity studies have not been conducted with ripretinib.
Genotoxicity
Ripretinib was found to be positive in an in vitro micronucleus assay. Ripretinib was not mutagenic in in vitro bacterial reverse mutation (Ames) assay nor in an in vivo rat bone marrow micronucleus assay, demonstrating the absence of significant genotoxic risk.
Reproductive and developmental toxicity
Dedicated fertility studies in male and female animals were not conducted with ripretinib. However, in a 13-week repeat-dose toxicity study in male rats, there were findings of degeneration in the seminiferous epithelium of the testes, and cellular debris of the epididymis in males administered 30 or 300 mg/kg/day but were considered of sufficient severity to affect reproduction at dose 300 mg/kg/day only (approximately 1.4 times the human exposure at 150 mg once daily).
In a pivotal embryofoetal development study, ripretinib was teratogenic in rats, inducing dose-related malformations primarily associated with the visceral and skeletal systems at a maternal dose of 20 mg/kg/day (approximately 1.0 times the human exposure at 150 mg once daily). Additionally, skeletal variations were already observed at 5 mg/kg/day. The developmental NOAEL for ripretinib was therefore established at 1 mg/kg/day (approximately 0.02 times the human exposure at 150 mg once daily).
A study investigating effects of ripretinib on the pre-/postnatal development was not performed.
Phototoxicity
Ripretinib indicates a potential for photoirritation/phototoxicity based on absorption in the UV visible range (above 290 nm). In vitro phototoxicity assessment in 3T3 mouse fibroblast cells suggest that ripretinib exhibits a potential for phototoxicity at clinically relevant concentrations following exposure to UVA and UVB radiation.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.