RANTUDIL FORTE Hard capsule Ref.[51125] Active ingredients: Acemetacin
Source: Medicines Authority (MT) Revision Year: 2023 Publisher: Viatris Healthcare Limited, Damastown Industrial Park, Mulhuddart, Dublin 15, DUBLIN, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Non-steroidal anti-inflammatory drugs and antirheumatics. Derivatives of acetic acid and related substances.
ATC code: M01AB11
Acemetacin belongs to the group of indoleacetic acid derivatives. Its effects can mainly be attributed to its metabolite indomethacin. Acemetacin is a non-steroidal antiphlogistic/analgesic agent which has demonstrated its efficacy in conventional experimental inflammatory animal models by inhibiting prostaglandin synthesis. In humans, acemetacin reduces pain caused by inflammation, swelling and fever. In addition, acemetacin inhibits ADP-induced platelet aggregation.
5.2. Pharmacokinetic properties
Acemetacin is rapidly and completely absorbed after oral administration of the usual entericcoated dosage forms. After repeated application (3 times daily up to 10 days), bioavailability reaches almost 100%.
Its biologically active metabolite is indometacin.
The following pharmacologically inactive metabolites were found: O-desmethyl, des-pchlorobenzoyl and O-desmethyl-des-p-chlorobenzoyl derivatives of acemetacin or indometacin as well as their glucuronide conjugates.
Depending on the duration of stomach passage, peak plasma levels of about 2 mg/l for acemetacin and 1.4 mg/l for its active metabolite indometacin are reached after 1–16 hours, on average after 2-3 hours.
About 50% of the active substance is eliminated with the faeces in a metabolised form. Following hepatic metabolic degradation (hydroxylation and conjugation), about 40% will be eliminated as pharmacologically inactive metabolites. Elimination half-time comes to about 4.5 hours and is mainly independent of hepatic or renal function. Plasma protein binding is high.
Acemetacin accumulates in the inflamed region. After six days of treatment and six hours following the last application, significantly higher concentrations of the active substance were found in synovial fluid, synovial membrane, musculature and bones than in the blood.
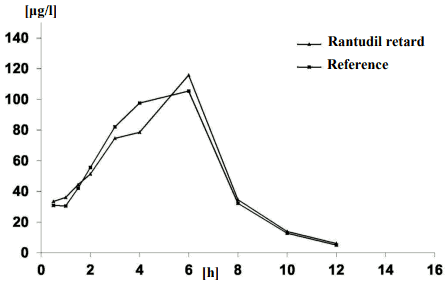
Bioavailability
In 1997, a bioavailability study on 28 volunteers for Rantudil retard hard capsules, sustainedrelease 90mg was carried out in comparison with a reference preparation:
| Rantudil retard | Reference preparation | |
|---|---|---|
| AUC0-14 (μg x h/l) | 673 (1.44) | 685 (1.44) |
| Cmax (μg/l) | 153 (1.61) | 163 (1.62) |
| tmax (h) | 6 (2-8) | 5 (1.5-8) |
| t1/2 (h) | 1.8 (1.33) | 1.9 (1.54) |
Values are given as geometric means and standard deviation (after single treatment).
Mean plasma concentrations in comparison with a reference preparation shown in a concentration-time diagram.
5.3. Preclinical safety data
In animal studies, sub-chronic and chronic toxicity of acemetacin primarily manifested as lesions and ulceration in the gastrointestinal tract, increased tendency to bleed, hepatic and renal lesions, as well as changes in blood count. The body weight-related no-effect doses were 1.0 mg/kg body weight in rats and 4.5 mg/kg body weight in monkeys above the human therapeutic range.
In vitro and in vivo mutagenicity tests did not reveal any mutagenic effect of acemetacin. In long-term studies in rats and mice, no indication of a carcinogenic potential of acemetacin was found.
The embryotoxic potential of acemetacin has been studied in two animal species (rats and rabbits). Foetal death and growth retardation were observed at doses in the maternal-toxic range. Malformations were not observed. Acemetacin prolonged gestation, as well as the duration of the birth process. No adverse effects on fertility were found.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.