RETSEVMO Hard capsule Ref.[28022] Active ingredients: Selpercatinib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Eli Lilly Nederland B.V., Papendorpseweg 83, 3528BJ Utrecht, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic and immunomodulating agents, antineoplastic agents, protein kinase inhibitors
ATC code: L01EX22
Mechanism of action
Selpercatinib is an inhibitor of the rearranged during transfection (RET) receptor tyrosine kinase. Selpercatinib inhibited wild-type RET and multiple mutated RET isoforms as well as VEGFR1 and VEGFR3 with IC50 values ranging from 0.92 nM to 67.8 nM. In other enzyme assays, selpercatinib also inhibited FGFR 1, 2, and 3 at higher concentrations that were still clinically achievable. In a binding assay at the concentration of 1 μM selpercatinib, significant antagonist binding activity (>50%) was observed for the 5-HT (serotonin) transporter (70.2% antagonist) and α2C adrenoreceptor (51.7% antagonist). The concentration of 1 μM is approximately 7-fold higher than the maximum unbound plasma concentration of at the efficacious dose of selpercatinib.
Certain point mutations in RET or chromosomal rearrangements involving in-frame fusions of RET with various partners can result in constitutively activated chimeric RET fusion proteins that can act as oncogenic drivers by promoting cell proliferation of tumour cell lines. In in vitro and in vivo tumour models, selpercatinib demonstrated anti-tumour activity in cells harboring constitutive activation of RET protein resulting from gene fusions and mutations, including CCDC6-RET, KIF5B-RET, RET V804M, and RET M918T. In addition, selpercatinib showed anti-tumour activity in mice intracranially implanted with a patient-derived RET fusion-positive tumour.
Pharmacodynamic properties
Cardiac electrophysiology
In a thorough QT study with positive control in 32 healthy subjects, no large change (that is, >20 ms) in the QTcF interval was detected at selpercatinib concentrations similar to those observed with a therapeutic dosing schedule. An exposure-response analysis indicated that supra therapeutic concentrations, could lead to an increase in QTc >20 ms. In patients receiving selpercatinib, QT interval prolongation was reported. Therefore, dose interruption or modification may be required in patients (see sections 4.2 and 4.4).
Clinical efficacy and safety
The efficacy of Retsevmo was evaluated in adult patients with advanced RET fusion-positive NSCLC, RET fusion-positive thyroid cancer, other RET fusion-positive solid tumours, and in adult and adolescent patients with RET-mutant MTC enrolled in a phase 1/2, multicenter, open-label, single-arm clinical study: Study LIBRETTO-001. Efficacy of Retsevmo in RET fusion-positive NSCLC was confirmed in the Phase 3 Study LIBRETTO-431 (see section Treatment-naïve RET fusion-positive NSCLC). Efficacy of Retsevmo in RET-mutant MTC was confirmed in the Phase 3 Study LIBRETTO-531 (see section Vandetanib and cabozantinib naïve RET-mutant medullary thyroid cancer (MTC)).
Study LIBRETTO-001 included two parts: phase 1 (dose escalation) and phase 2 (dose expansion). The primary objective of the phase 1 portion was to determine the recommended phase 2 dose of selpercatinib. The primary objective of the phase 2 part was to evaluate the anti-tumour activity of selpercatinib by determining ORR, as assessed by independent review committee. Patients with measurable or non-measurable disease as determined by RECIST 1.1, with evidence of a RET gene alteration in tumour were enrolled. Patients with CNS metastases were eligible if stable, while patients with symptomatic primary CNS tumour, metastases, leptomeningeal carcinomatosis or spinal cord compression were excluded. Patients with known primary driver alteration other than RET, clinically significant active cardiovascular disease or history of myocardial infarction, QTcF interval >470 msec were excluded.
Patients in the phase 2 portion of the study received Retsevmo 160 mg orally twice daily until unacceptable toxicity or disease progression. Identification of a RET gene alteration was prospectively determined in local laboratories using next generation sequencing (NGS), polymerase chain reaction (PCR), or fluorescence in situ hybridization (FISH). The primary efficacy outcome measure was objective response rate (ORR) according to RECIST v1.1 as evaluated by a Blinded Independent Review Committee (IRC). Secondary efficacy outcomes included duration of response (DOR), progression free survival (PFS) and overall survival (OS).
Treatment-naïve RET fusion-positive NSCLC
LIBRETTO-431
The efficacy of Retsevmo in RET fusion-positive NSCLC was confirmed in LIBRETTO-431, a phase 3 multicentre, randomised, open-label comparator study, comparing selpercatinib to platinum-based and pemetrexed therapy with or without pembrolizumab in patients with advanced or metastatic RET fusion-positive NSCLC. Adult patients with histologically confirmed, unresectable, locally advanced or metastatic NSCLC with no previous systemic therapy for metastatic disease were eligible. Patients who received adjuvant or neoadjuvant therapy if the last dose of systemic treatment was completed at least 6 months prior to randomisation were also eligible. Patients received 160 mg of selpercatinib twice daily (starting dose) or platinum-based and pemetrexed therapy with or without pembrolizumab. Patients were stratified according to geographic region (East Asia vs. elsewhere), status with respect to investigator assessed brain metastases at baseline (absent or unknown vs present), and whether the investigator had intended (before randomisation) to treat the patient with or without pembrolizumab. The primary efficacy outcome measure was PFS per RECIST 1.1 by BICR. Secondary efficacy outcomes included OS, ORR/DOR/DCR by BICR, intracranial ORR/DOR by BICR, and time to deterioration in pulmonary symptoms by NSCLC-SAQ.
Of the 261 patients enrolled and randomized in Study LIBRETTO-431 intention to treat (ITT) population, 212 were stratified according to whether the investigator would intend for the patient to receive pembrolizumab (before randomisation), to form the ITT-Pembrolizumab population. In the ITT-Pembrolizumab population, 129 patients received selpercatinib while 83 received platinum-based pemetrexed chemotherapy with pembrolizumab. The median age of patients in the ITT-Pembrolizumab population was 61.5 years (range 31 to 84 years). 53.3% of patients were female. 41.3% of patients were White, 56.3% were Asian, 1% were Black. 67.9% were never smokers. In the ITT Pembrolizumab population, 93% had metastatic disease, and 20.3% of patients had CNS metastases at baseline. ECOG performance status was reported as 0-1 (96.7%) or 2 (3.3%). The most common fusion partner was KIF5B (44.8%), followed by CCDC6 (9.9%). The study met its primary endpoint of improving PFS in both the ITT-Pembrolizumab and ITT populations. Primary efficacy results for the ITT-Pembrolizumab population for treatment naïve patients with RET fusion-positive NSCLC are summarised in Table 4 and Figure 1.
Table 4. LIBRETTO-431: Summary of efficacy data (BICR assessment, ITT-Pembrolizumab population):
| Selpercatinib | Control (platinum- based pemetrexed chemotherapy with pembrolizumab) | |
|---|---|---|
| Progression-free survival | N=129 | N=83 |
| Median [months] (95% CI) | 24.84 (16.89, NE) | 11.17 (8.77, 16.76) |
| Hazard ratio (95% CI) | 0.465 (0.309, 0.699) | |
| Stratified log rank p-value | 0.0002 | |
| 24 months PFS rate () (95 CI) | 54.2 (43.6, 63.6) | 31.6 (20.1, 43.7) |
| Objective response (CR + PR) | ||
| % (95% CI) | 83.7 (76.2, 89.6) | 65.1 (53.8, 75.2) |
| Complete response n (%) | 9 (7.0) | 5 (6.0) |
| Partial response n (%) | 99 (76.7) | 49 (59.0) |
| Duration of response* | ||
| Median [months] (95% CI) | 24.18 (17.94, NE) | 11.47 (9.66, 23.26) |
| Rate (%) of patients with duration of response | ||
| 24 months (95% CI) | 59.6 (47.5, 69.8) | 22.8 (6.3, 45.5) |
NE = not estimable
Figure 1. LIBRETTO-431: Kaplan-Meier plot of progression-free survival (BICR assessment, ITT-Pembrolizumab population):
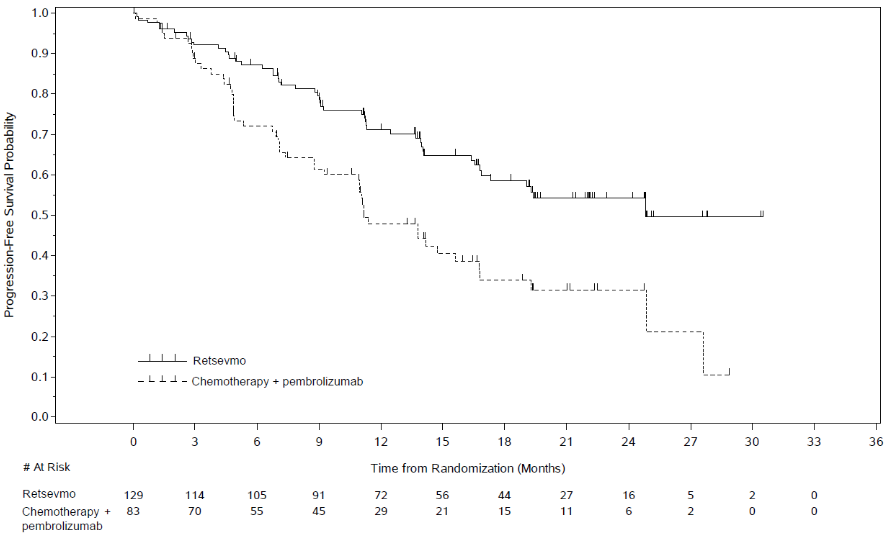
Data Cut-off date: 01 May 2023
OS was not mature at the time of the primary PFS analysis (40 events observed across the two arms). The censoring rate was 80.6% in the selpercatinib arm and 81.9% in the control arm and results may be confounded due to cross over. In the ITT-Pembrolizumab population the OS HR was 0.961 ([95% CI: 0.503, 1.835]; p=0.9033).
In the ITT-Pembrolizumab population, selpercatinib significantly delayed time to worsening of patient-reported NSCLC symptoms, as measured by the NSCLC-Symptom Assessment Questionnaire total score (≥2-point increase) compared with the control (HR: 0.34 [95% CI: 0.20, 0.55]; median time was not reached for selpercatinib arm versus 1.9 months [95% CI: 0.7, 6.6]) for the control arm. In addition, selpercatinib significantly delayed time to confirmed deterioration of physical function and maintained overall quality of life over time.
LIBRETTO-001
Of the 362 RET fusion-positive NSCLC patients enrolled in LIBRETTO-001, 69 were treatment naïve. The median age was 63 years (range 23 years to 92 years). 62.3% of patients were female. 69.6% of patients were White, 18.8% were Asian, 5.8% were Black and 69.6% were never smokers. Most patients (98.6%) had metastatic disease at enrolment and 23.2% had CNS metastasis at baseline as assessed by investigator. ECOG performance status was reported as 0-1 (94.2%) or 2 (5.8%). The most common fusion partner was KIF5B (69.6%), followed by CCDC6 (14.5%) and then NCOA4 (1.4%). Efficacy results for treatment-naive RET fusion-positive NSCLC patients are summarised in Table 5.
Table 5. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 69 |
| Objective response (CR + PR) | |
| % (95% CI) | 82.6 (71.6, 90.7) |
| Complete response n (%) | 5 (7.2) |
| Partial response n (%) | 52 (75.4) |
| Duration of response (months)* | |
| Median, 95% CI | 20.23 (15.4, 29.5) |
| Rate (%) of patients with duration of response | |
| ≥6 months (95% CI) | 87.5 (75.5, 93.8) |
| ≥12 months (95% CI) | 66.7 (52.4, 77.6) |
* Median duration of follow-up was 37.09 months (25th, 75th percentile: 24.0, 45.1)
Data Cut-off date: 13 January 2023.
Previously treated RET fusion-positive NSCLC
A total of 247 patients had received prior platinum-based chemotherapy in Study LIBRETTO-001. The median age was 61 years (range 23 years to 81 years). 56.7% of patients were female. 43.7% of patients were White, 47.8% were Asian, 4.9% were Black, and 66.8% were never smokers. Most patients (98.8%) had metastatic disease at enrolment and 31.2% had CNS metastasis at baseline as assessed by investigator. ECOG performance status was reported as 0-1 (97.1%) or 2 (2.8%). The most common fusion partner was KIF5B (61.9%), followed by CCDC6 (21.5%) and then NCOA4 (2.0%). The median number of prior systemic therapies was 2 (range 1–15) and 43.3% (n=107/247) received 3 or more prior systemic regimens; prior treatments included anti PD1/PD-L1 therapy (58.3%), multi-kinase inhibitor (MKI) (31.6%) and taxanes (34.8%); 41.3% had other systemic therapy. Efficacy results for previously treated RET fusion-positive NSCLC patients are summarised in Table 6.
Table 6. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 247 |
| Objective response (CR + PR) | |
| % (95% CI) | 61.5 (55.2, 67.6) |
| Complete response n (%) | 20 (8.1) |
| Partial response n (%) | 132 (53.4) |
| Duration of response (months)* | |
| Median (95% CI) | 31.6 (20.4, 42.3) |
| Rate (%) of patients with duration of response | |
| ≥6 months (95% CI) | 87.0 (80.4, 91.5) |
| ≥12 months (95% CI) | 73.0 (65.0, 79.5) |
* Median duration of follow-up was 39.52 months (25th, 75th percentile: 24.6, 45.0)
Data Cut-off date: 13 January 2023.
CNS response in RET fusion-positive NSCLC
In Study LIBRETTO-431 the CNS ORR assessed by BICR was 82.4% (14/17 95% CI: 56.6, 96.2) in the 17 patients with measurable brain metastases at baseline treated with selpercatinib, versus 58.3% (7/12 95% CI: 27.7 to 84.4) in the 12 patients in the control arm of the ITT-Pembrolizumab population. CR was observed in 6/17 (35.3%) of patients in the selpercatinib arm versus 2/12 (16.7%) patients in the control arm. With a median follow up time for DOR of 9.92 months (95% CI: 7.66, 18.10) in the selpercatinib arm and 12.68 months (95% CI: 2.79, NE) in the control arm, the median DOR was not reached for selpercatinib (95% CI: 7.62, NE) compared to 13.4 months (95% CI: 3.45, NE) with control. In 192 patients with intracranial baseline scans available, the cause-specific hazard ratio for the time to CNS progression, as assessed by BICR, was 0.28; 95% CI: 0.12, 0.68 (HR of 0.17; 95% CI: 0.04, 0.69 for 150 patients without baseline intracranial metastases, and HR of 0.61; 95% CI: 0.19, 1.92 for 42 patients with baseline intracranial metastases). 8 patients (6.7%) in the selpercatinib arm had a first event of CNS progression compared to 13 patients (18.1%) in the control arm.
The CNS ORR assessed by IRC was 84.6% (22/26; 95% CI: 65.1, 95.6) in 26 patients with measurable disease in Study LIBRETTO-001. CR was observed in 7 (26.9%) patients and PR in 15 (57.5%) patients. The median CNS DOR was 9.36 months (95% CI: 7.4, 15.3).
Systemic treatment-naive RET fusion-positive thyroid cancer
Of the RET fusion-positive thyroid cancer patients naive to systemic therapy other than radioactive iodine, and enrolled in LIBRETTO-001, 24 patients had the opportunity to be followed for at least 6 months and were considered efficacy eligible. The median age was 60.5 years (range 20 to 84 years). 58.3% of patients were male. 75% of patients were White. ECOG performance status was reported as 0-1 (95.8%) or 2 (4.2%). 100% of patients had a history of metastatic disease. 22 out of the 24 patients (91.7%) received radioactive iodine prior to enrolment and therefore were considered radioactive iodine refractory. The different histologies represented in the 24 patients included: papillary (n=23) and poorly differentiated (n=1). The most common fusion partner was CCDC6 (62.5%) followed by NCOA4 (29.2%). Efficacy results for systemic treatment-naive RET fusion-positive thyroid cancer patients are summarised in Table 7.
Table 7. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 24 |
| Objective response (CR + PR) | |
| % (95% CI) | 95.8 (78.9, 99.9) |
| Complete response n (%) | 5 (20.8) |
| Partial response n (%) | 18 (75.0) |
| Duration of response (months)* | |
| Median (95% CI) | NE (42.8, NE) |
| Rate (%) of patients with duration of response | |
| ≥12 months (95% CI) | 100.0 (NE, NE) |
| ≥24 months (95% CI) | 90.9 (50.8, 98.7) |
NE = not estimable
* Median duration of follow-up was 17.81 months (25th, 75th percentile: 9.2, 42.3)
Data Cut-off date: 13 January 2023
Previously treated RET fusion-positive thyroid cancer
Of the RET fusion-positive thyroid cancer patients previously treated with systemic therapy other than Radioactive iodine, and enrolled in LIBRETTO-001, 41 patients had the opportunity to be followed for at least 6 months and were considered efficacy eligible. The median age was 58 years (range 25 to 88 years). 43.9% of patients were male. 58.5% of patients were White while 29.3% were Asian and 7.3% were Black. ECOG performance status was reported as 0-1 (92.7%) or 2 (7.3%). 100% of patients had metastatic disease. Patients had received a median of 3 prior systemic therapies (range: 1-7). The most common prior therapies included radioactive iodine (73.2%), MKI (85.4%). 9.8% had other systemic therapy. The different histologies represented in the 41 patients included: papillary (n=31), poorly differentiated (n = 5), anaplastic (n = 4), and Hurthle cell (n=1). The most common fusion partner was CCDC6 (61.0%) followed by NCOA4 (19.5%).
Efficacy results for previously treated RET fusion-positive thyroid cancer are summarised in Table 8.
Table 8. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 41 |
| Objective response (CR + PR) | |
| % (95% CI) | 85.4 (70.8, 94.4) |
| Complete response n (%) | 5 (12.2) |
| Partial response n (%) | 30 (73.2) |
| Duration of response (months)* | |
| Median (95% CI) | 26.7 (12.1, NE) |
| Rate (%) of patients with duration of response | |
| ≥12 months (95% CI) | 71.7 (52.4, 84.2) |
| ≥24 months (95% CI) | 50.7 (30.4, 67.8) |
NE = not estimable
* Median duration of follow-up was 33.87 months (25th, 75th percentile: 12.9, 44.8)
Data Cut-off date: 13 January 2023.
Vandetanib and cabozantinib naïve RET-mutant medullary thyroid cancer (MTC)
LIBRETTO-531
The efficacy of Retsevmo in RET-mutant MTC was confirmed in LIBRETTO-531, a phase 3 multicenter, randomised, open-label comparator study, comparing selpercatinib to physician’s choice of cabozantinib or vandetanib in patients with progressive, advanced, kinase inhibitor naïve, RET-mutant MTC. Adult or adolescent patients with histologically confirmed, unresectable, locally advanced, or metastatic MTC with no previous treatment with a kinase inhibitor were eligible. Patients received 160 mg of selpercatinib twice daily (starting dose) or physician’s choice of cabozantinib (140 mg once daily) or vandetanib (300 mg once daily). Patients were stratified according to RET mutation (M918T vs. other), and the intended treatment if randomised to control arm (cabozantinib vs vandetanib). The primary efficacy outcome measure was PFS per RECIST 1.1 by BICR. Key secondary efficacy outcomes included treatment failure-free survival (TFFS) and comparative tolerability, and other secondary efficacy outcomes included OS and ORR/DOR by BICR.
Of the 291 patients enrolled and randomised in LIBRETTO-531 to form the ITT population, 193 were randomised to the selpercatinib arm, and 98 were randomised to the control arm. Of the 98 patients randomised to the control arm, 73 were stratified to cabozantinib, and 25 were stratified to vandetanib. The median age of patients in the ITT population was 55 years (range: 12 to 84 years). 37.1% of patients were female. 69.4% of patients were White, 27.7% were Asian, 2.9% were Black. Most patients (77%) had metastatic disease at enrolment. ECOG performance status was reported as 0-1 (98.3%) or 2 (1%). The most common mutation was M918T (62.5%). The study met its primary endpoint of improving PFS in the ITT population. Efficacy results for the ITT population are summarized in Table 9 and Figure 2.
Table 9. LIBRETTO-531: Summary of efficacy data (BICR assessment, ITT population):
| Selpercatinib | Control (Cabozantinib or Vandetanib) | |
|---|---|---|
| Progression-free survival | N=193 | N=98 |
| Median [months] (95% CI) | NE (NE, NE) | 16.76 (12.22, 25.10) |
| Hazard ratio (95% CI) | 0.280 (0.165, 0.475) | |
| Stratified log rank p-value | <0.0001 | |
| 30 months PFS rate () 95 CI | 76.4 (66.5, 83.8) | 24.8 (6.9, 48.3) |
| Treatment failure-free survival* | N=193 | N=98 |
| Median [months] (95% CI) | NE (NE, NE) | 13.93 (11.27, 25.10) |
| Hazard ratio (95% CI) | 0.254 (0.153, 0.423) | |
| Stratified log rank p-value | <0.0001 | |
| 30 months TFFS rate () 95 CI | 75.8 (65.9, 83.2) | 25.3 (7.2, 48.8) |
| Objective response (CR + PR) | ||
| % (95% CI) | 69.4 (62.4, 75.8) | 38.8 (29.1, 49.2) |
| Complete response n (%) | 23 (11.9) | 4 (4.1) |
| Partial response n (%) | 111 (57.5) | 34 (34.7) |
| Duration of response# | ||
| Median [months] (95% CI) | NE (NE, NE) | 16.56 (10.41, NE) |
| Rate (%) of patients with duration of response | ||
| ≥24 months (95% CI) | 79.1 (66.9, 87.2) | NE (NE, NE) |
NE = not estimable
* Treatment failure-free survival is defined as the time from randomization to the first occurrence of: documented radiographic disease progression per RECIST 1.1, or unacceptable toxicity leading to treatment discontinuation as assessed by the investigator, or death due to any cause.
# Median duration of follow-up was 11.14 months (25th, 75th percentile: 5.62, 16.62) in the selpercatinib arm and 12.81 months (25th and 75th percentile: 6.34, 15.51) in the control arm.
Data Cut-off date: 22 May 2023
Figure 2. LIBRETTO-531: Kaplan-Meier plot of progression-free survival (BICR assessment, ITT population):
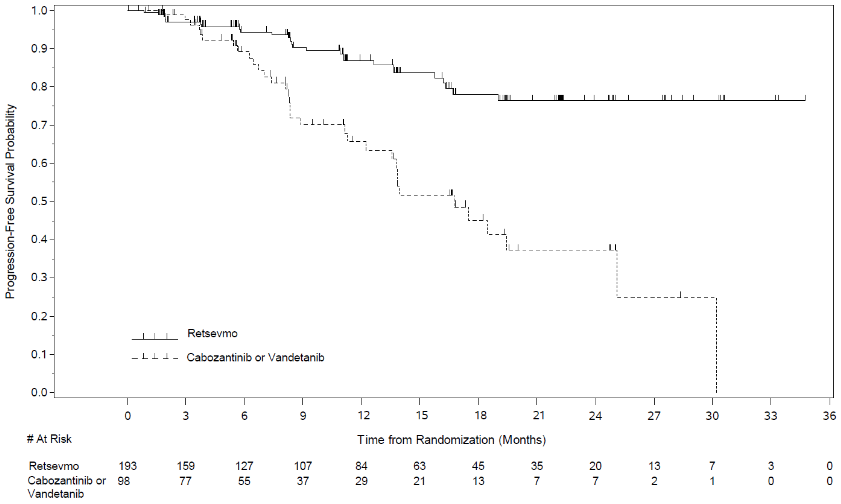
Data cut off: 22 May 2023
At the time of the primary PFS analysis, 18 OS events were observed across the two arms. In the ITT population, the OS HR was 0.374 ([95% CI: 0.147, 0.949]). The censoring rate was 95.9% in the selpercatinib arm and 89.8% in the control arm. Comparative tolerability was evaluated in 242 patients (selpercatinib arm, N=161; control arm, N=81). The selpercatinib arm had a statistically significantly lower proportion of time on treatment where patients reported “high side effect bother” (8%) than the control arm (24%) (95% CI: -23%, -10%, p<0.0001) as assessed by Functional Assessment of Cancer Therapy item GP5 response 3 “Quite a bit” or 4 “Very much”.
At a later OS analysis, with a data lock of 11 March 2024, 26 events were observed across the two arms and the HR was 0.275 (95% CI: 0.124, 0.608). The PFS HR for this analysis was 0.202 (95% CI: 0.128, 0.320) and the ORR for selpercatinib was 82.4% compared to 43.9% for the control arm.
LIBRETTO-001
Of the 324 RET-mutant MTC patients enrolled in LIBRETTO-001, 143 were naïve to treatment with cabozantinib and vandetanib. Of these 116 were treatment naïve to other systemic therapy and 27 had previously received other systemic therapy. Among patients naïve to cabozantinib and vandetanib, the median age was 57 years (range 15 to 87 years). 2 patients (1.4%) were <18 years of age. 58.0% of patients were male. 86.7% of patients were White, 5.6% were Asian, 1.4% were Black. Most patients (97.9%) had metastatic disease at enrolment. ECOG performance status was reported as 0-1 (95.9%) or 2 (4.2%). The most common mutation was M918T (60.1%), followed by extracellular cysteine mutations (23.8%). Efficacy results for cabozantinib and vandetanib treatment-naive RET-mutant MTC patients are summarised in Table 10.
Table 10. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 143 |
| Objective response (CR + PR) | |
| % (95% CI) | 82.5 (75.3, 88.4) |
| Complete response n (%) | 34 (23.8) |
| Partial response n (%) | 84 (58.7) |
| Duration of response (months)* | |
| Median, 95% CI | NE (51.3, NE) |
| Rate (%) of duration of response | |
| ≥12 months (95% CI) | 91.4 (84.6, 95.3) |
| ≥24 months (95% CI) | 84.1 (75.9, 89.7) |
NE = not estimable
* Median duration of follow-up was 39.4 months (25th, 75th percentile: 32.3, 45.4).
Data cut-off date 13 January 2023.
Previously treated RET-mutant medullary thyroid cancer
Of the RET-mutant MTC patients enrolled in LIBRETTO-001, 152 were previously treated with cabozantinib and/or vandetanib, and considered efficacy eligible. The median age was 58 years (range 17 years to 90 years); 1 patient (0.7%) was <18 years of age. 63.8% of patients were male. 90.1% of patients were White while 1.3% were Asian, and 1.3% were Black. ECOG performance status was reported as 0-1 (92.7%) or 2 (7.2%). 98.0% of patients had metastatic disease. The most common mutation was M918T (65.1%), followed by extracellular cysteine mutations (15.8%). 100% (n=152) of patients received prior systemic therapy with a median of 2 prior systemic regimens and 27.6% (n=42) received 3 or more prior systemic regimens.
Efficacy results for previously treated RET-mutant MTC are summarised in Table 11.
Table 11. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 152 |
| Objective response (CR + PR) | |
| % (95% CI) | 77.6 (70.2, 84.0) |
| Complete response n (%) | 19 (12.5) |
| Partial response n (%) | 99 (65.1) |
| Duration of response (months)* | |
| Median (95% CI) | 45.3 (33.6, NE) |
| Rate (%) of duration of response | |
| ≥12 months (95% CI) | 83.0 (74.6, 88.8) |
| ≥24 months (95% CI) | 66.4 (56.3, 74.7) |
NE = not estimable
* Median duration of follow-up was 38.3 months (25th, 75th percentile: 23.0, 46.1).
Data cut-off date 13 January 2023.
Other RET Fusion-Positive Solid Tumours
Efficacy was evaluated in 52 patients with RET fusion-positive tumours other than NSCLC and thyroid cancer with disease progression on or following prior systemic treatment or who had no satisfactory alternative treatment options. The median age was 54 years (range 21 to 85); 51.9% were female; 67.3% were White, 25.0% were Asian, and 5.8% were Black; ECOG performance status was 0-1 (92.3%) or 2 (7.7%) and 96.2% of patients had metastatic disease. Forty-seven patients (90.4%) received prior systemic therapy with a median of 2 prior systemic therapies (range 0 to 9) and 28.8% received 3 or more prior systemic therapies. No patients had been previously treated with a selective RET inhibitor. The most common cancers were pancreatic (25%), colon (25%), and salivary (7.7%). The most common fusion partners were NCOA4 (34.6%), CCDC6 (17.3%), and KIF5B (11.5%). Efficacy results for RET fusion-positive solid tumours other than NSCLC and thyroid cancer are summarized in Table 12 and Table 13.
Table 12. LIBRETTO-001: Objective response and duration of response:
| Efficacy eligible patients IRC assessment | |
|---|---|
| N | 52 |
| Objective response (CR+PR) | |
| % (95% CI) | 44.2 (30.5, 58.7) |
| Complete response n (%) | 3 (5.8) |
| Partial response n (%) | 20 (38.5) |
| Duration of Response (months)* | |
| Median (95% CI) | 37.19 (13.3, NE) |
| Rate (%) of duration of response | |
| ≥6 months (95% CI) | 84.7 (59.5, 94.8) |
| ≥12 months (95% CI) | 79 (53.1, 91.6) |
* Median duration of follow-up was 28.55 months (25th, 75th percentile: 11.2, 40.9)
NE = not estimable
Data cut-off date 13 January 2023
Table 13. LIBRETTO-001: Objective response and duration of response by Tumour Type:
| Tumour Type | Patients (N=52) | ORR (IRC assessment) | DOR Range (months) | |
|---|---|---|---|---|
| n (%) | 95% CI | |||
| Pancreatic | 13 | 7 (53.8) | 25.1, 80.8 | 2.50, 52.14 |
| Colorectal | 13 | 4 (30.8) | 9.1, 61.4 | 1.84+, 13.31 |
| Salivary | 4 | 2 (50.0) | 6.8, 93.2 | 5.72, 37.19 |
| Cholangiocarcinoma | 3 | 1 (33.3) | 0.8, 90.6 | 14.82 |
| Unknown primary | 3 | 1 (33.3) | 0.8, 90.6 | 9.23 |
| Sarcoma | 3 | 1 (33.3) | 0.8, 90.6 | 31.44+ |
| Breast | 2 | PR, CR | NA | 2.30+, 17.28 |
| Xanthogranuloma | 2 | NE, NEa | NA | NA |
| Carcinoma of the skin | 2 | NE, PR | NA | 14.82+ |
| Carcinoid | 1 | PR | NA | 40.94+ |
| Ovarian | 1 | PR | NA | 28.55+ |
| Pulmonary carcinosarcoma | 1 | NE | NA | NA |
| Rectal neuroendocrine | 1 | NE | NA | NA |
| Small intestine | 1 | CR | NA | 24.54 |
| Neuroendocrine | 1 | PR | NA | 3.54+ |
| Small cell lung cancer | 1 | SD | NA | NA |
+ denotes ongoing response.
a One xanthogranuloma patient had disease that could not be evaluated by IRC due to skin being the only site of disease. Based on investigator assessment, this patient had a CR.
CI = confidence interval, CR = complete response, DOR = duration of response, NA = not applicable, NE = not evaluable, ORR = objective response rate, PR = partial response, SD = stable disease.
Data cut-off date 13 January 2023
Due to the rarity of RET fusion-positive cancer, patients were studied across multiple tumour types with a limited number of patients in some tumour types, causing uncertainty in the ORR estimate per tumour type. The ORR in the total population may not reflect the expected response in a specific tumour type.
Paediatric population
As of 13 January 2023, 10 patients with RET fusion-positive thyroid cancer aged 12 to ≤21 years have been treated in LIBRETTO-121, an ongoing Phase ½ study in paediatric patients with an advanced solid or primary CNS tumour harbouring an activating RET alteration. Of these 10 patients, 8 patients were less than 18 years of age. Of the 10 patients, 4 were previously treated with radioactive iodine only, 2 had received prior systemic therapy that did not include radioactive iodine and 4 were not previously treated with any systemic therapy. For all 10 patients, per IRC, objective response rate was 60.0% (95% CI: 26.2, 87.8). 3 patients had confirmed complete response whilst 3 patients had confirmed partial response.
The European Medicines Agency has waived the obligation to submit the results of studies with selpercatinib in patients aged 6 months and below in solid tumours (see section 4.2 for information on paediatric use).
The European Medicines Agency has deferred the obligation to submit the results of studies with selpercatinib in one or more subsets of the paediatric population in relapsed/refractory solid tumours, including RET fusion-positive solid tumours, RET-mutant medullary thyroid cancer, and other tumours with RET alteration/activation (see section 4.2 for information on paediatric use).
Conditional approval
This medicinal product has been authorised under a so-called ‘conditional approval’ scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
The pharmacokinetics of selpercatinib were evaluated in patients with locally advanced or metastatic solid tumours administered 160 mg twice daily unless otherwise specified. Steady-state selpercatinib AUC and Cmax increased in a linear to supra-dose proportional manner over the dose range of 20 mg once daily to 240 mg twice daily.
Steady-state was reached by approximately 7 days and the median accumulation ratio after administration of 160 mg twice daily was 3.4-fold. Mean steady-state selpercatinib [coefficient of variation (CV%)] Cmax was 2,980 (53%) ng/mL and AUC0-24h was 51,600 (58%) ng*h/mL.
In vivo studies indicate that selpercatinib is a mild inhibitor of P-gp.
In vitro studies indicate that selpercatinib does not inhibit or induce CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6 at clinically relevant concentrations.
In vitro studies indicate that selpercatinib inhibits MATE1, and BCRP, but does not inhibit OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP, and MATE2-K at clinically relevant concentrations. Selpercatinib may increase serum creatinine by decreasing renal tubular secretion of creatinine via inhibition of MATE1.
Absorption
After an oral dose of 160 mg, Retsevmo was rapidly absorbed, with Tmax of approximately 2 hours. Geometric mean absolute oral bioavailability was 73.2% (range: 60.2-81.5%).
Effect of food
Compared to selpercatinib AUC and Cmax in the fasted state, selpercatinib AUC was increased by 9% and Cmax was reduced by 14% after oral administration of a single 160 mg dose to healthy subjects taken with a high-fat meal. These changes were not considered to be clinically relevant. Therefore, selpercatinib can be taken with or without food.
Distribution
Selpercatinib mean (CV%) volume of distribution (Vss/F), estimated by Population PK analysis, is 203.1 (69%) L following oral administration of selpercatinib in adult patients. Selpercatinib is 96% bound to human plasma proteins in vitro and binding is independent of concentration. The blood-to-plasma concentration ratio is 0.7.
Biotransformation
Selpercatinib is metabolized predominantly by CYP3A4. Following oral administration of a single [14C] radiolabeled 160 mg dose of selpercatinib to healthy subjects, unchanged selpercatinib constituted 86% of the measured radioactive components in plasma.
Elimination
The mean (CV%) clearance (CL/F) of selpercatinib is 5.5 (45%) L/h and the half-life is 26.5 hours following oral administration of selpercatinib in adult patients. Following oral administration of a single [14C] radiolabeled 160 mg dose of selpercatinib to healthy subjects, 69% (14% unchanged) of the administered radioactivity was recovered in faeces and 24% (11.5% unchanged) was recovered in urine.
Special populations
Age, gender and body weight
Age (range: 12 years to 92 years) or gender had no clinically meaningful effect on the pharmacokinetics of Retsevmo. Patients with a body weight < 50 kg should start Retsevmo treatment with a dose of 120 mg twice daily, while patients ≥ 50 kg should start Retsevmo treatment with a dose of 160 mg twice daily.
Hepatic impairment
Selpercatinib AUC0-∞ increased by 7% in subjects with mild, 32% in subjects with moderate Child-Pugh classification. Thus, selpercatinib exposure (AUC) in subjects with mild and moderate hepatic impairment (Child-Pugh class A and B) is comparable to exposure in healthy subjects when a dose of 160 mg is administered. Selpercatinib AUC0-∞ increased by 77% in subjects with severe hepatic impairment (Child-Pugh class C). There is limited clinical data on the safety of selpercatinib in patients with severe hepatic impairment. Therefore, dose modification is recommended for patients with severe hepatic impairment (section 4.2).
Renal impairment
In a clinical pharmacology study using single dose selpercatinib 160 mg, exposure (AUC) was unchanged in subjects with mild, moderate, or severe renal impairment. End stage renal disease (eGFR <15 ml/min) and dialysis patients have not been studied.
Paediatric population
Based on limited pharmacokinetic data, the Cmax and AUC was similar in adolescent patients, 12-18 years of age, and in adults.
5.3. Preclinical safety data
Repeat-dose studies were conducted in juvenile and adolescent/adult rats and adolescent/adult minipigs to characterize toxicity. Target organs of toxicity common to the rat and minipig were hematopoietic system, lymphoid tissues, tongue, pancreas, gastro-intestinal tract, epiphyseal growth plate, and male reproductive tissues. In general, toxicities in these organs were reversible; the exceptions were the testicular toxicity in adolescent/adult and juvenile animals, and changes in growth plates in juvenile rats. Reversible toxicity was observed in the ovaries in minipigs only. At high doses, gastrointestinal toxicity caused morbidity at exposures in minipigs that were generally lower than exposures determined in humans at the recommended dose. In one minipig study, females exhibited a slight, reversible increase in QTc prolongation of approximately 12% compared to controls and 7% compared to pre-dose values. Target organs of toxicity observed only in rats were incisor tooth, liver, vagina, lungs, Brunner’s gland, and multi-tissue mineralization associated with hyperphosphatemia. These toxicities only occurring in these organs in rats were reversible.
Juvenile toxicity
Selpercatinib exposure approximately 0.5-2 times the exposure in adult humans caused mortality in rats younger than 21 days old. Comparable exposure was tolerated in rats aged 21 days and older.
Juvenile and adolescent/adult rats and adolescent/adult minipigs with open growth plates administered selpercatinib exhibited microscopic changes of hypertrophy, hyperplasia, and dysplasia of growth plate cartilage (physis). In juvenile rats, the dysplasia at the growth plates was irreversible and associated with decreased femur length and reductions in bone mineral density. Skeletal changes were observed at exposure levels equivalent to those seen in adult patients taking the recommended dose of 160 mg BID.
Juvenile male rats administered selpercatinib and allowed to reach reproductive age after cessation of administration, exhibited decreased reproductive performance when mated with untreated female rats. Decreased fertility and copulation indices, increased pre- and post-implantation losses, and decreased number of viable embryos, were observed at an exposure approximately 3.4 times the efficacious exposure in adults.
Genotoxicity
Selpercatinib is not genotoxic at therapeutic doses. In an in vivo micronucleus assay in rats, selpercatinib was positive at concentrations >7 times the Cmax at the human dose of 160 mg twice daily. In an in vitro micronucleus assay in human peripheral blood lymphocytes, an equivocal response was observed at a concentration approximately 485 times the Cmax at the human dose.
Mutagenesis
Selpercatinib did not cause mutations in a bacterial mutagenicity assay.
Carcinogenesis
Long-term studies to assess the carcinogenic potential of selpercatinib have not been performed.
Embryotoxicitiy/Teratogenicity
Based on data from animal reproduction studies and its mechanism of action, selpercatinib can cause foetal harm when administered to a pregnant woman. Administration of selpercatinib to pregnant rats during organogenesis at maternal exposures that were approximately equal to those observed at the recommended human dose of 160 mg twice daily resulted in embryolethality and malformations.
Reproduction toxicity
Results of studies conducted in rats and minipigs suggest that selpercatinib could impair fertility in males and females.
In a fertility study in male rats, dose-dependent germ cell depletion and spermatid retention were observed at subclinical AUC-based exposure levels (0.2 times the clinical exposure at the recommended human dose). These effects were associated with reduced organ weights, reduced sperm motility, and an increase in the number of abnormal sperm at AUC-based exposure levels approximately twice the clinical exposure at the recommended human dose. Microscopic findings in the fertility study in male rats were consistent with effects in repeat dose studies in rats and minipigs, in which dose-dependent, non-reversible testicular degeneration was associated with reduced luminal sperm in the epididymis at subclinical AUC-based exposure levels (0.1 to 0.4 times the clinical exposure at the recommended human dose).
In a fertility and early embryonic study in female rats, a reduction in the number of estrous cycles as well as embryolethality were observed at AUC-based exposure levels approximately equal to clinical exposure at the recommended human dose. In repeat-dose studies in rats, reversible vaginal mucification with individual cell cornification and altered estrous cycles were noted at clinically relevant AUC-based exposure levels. In minipigs, decreased corpora lutea and/or corpora luteal cysts were observed at subclinical AUC-based clinical exposure levels (0.07 to 0.3 times the clinical exposure at the recommended human dose).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.